Oligodeoxynucleotides containing α-L-ribo configured LNA-type C-aryl nucleotides†
Received 5th September 2003, Accepted 24th October 2003
First published on 1st December 2003
Abstract
Synthesis of 2′-O,4′-C-methylene-α-L-ribofuranosyl derivatives containing phenyl and 1-pyrenyl aglycons, i.e., novel α-L-ribo configured LNA-type C-aryl nucleosides, has been accomplished. Key synthetic steps included stereoselective Grignard reactions on tetrahydrofuran aldehyde 12, configurational inversion of the resulting alcohol 13 into alcohol 15, and concomitant Mitsonobu cyclization furnishing the desired bicyclic furanosyl skeleton with a locked conformation. The phosphoramidite derivatives 19a and 19b were used for automated synthesis of 9-mer DNA and α-L-LNA oligonucleotides containing the α-L-LNA-type C-aryl monomers αLPhL and αLPyL containing a phenyl and pyrenyl aglycon, respectively. Thermal denaturation studies showed universal base pairing behavior for the pyrenyl monomer αLPyL when incorporated into a DNA or an α-L-LNA oligonucleotide.
Introduction
An oligonucleotide containing a universal base monomer binds isoenergetically with the four oligonucleotide complements containing each of the natural nucleotides opposite to the universal base analogue.1 Interesting universal bases include 2′-deoxy-β-D-ribofuranosyl derivatives of 3-nitropyrrole,2 5-nitroindole,3 isocarbostyril4 and 8-aza-7-deazaadenine.5 Also pyrenyl derivatives6–8 of 2′-deoxy-β-D-ribofuranose and LNA,9§ have been reported as universal bases.Recently, we have reported universal base pairing behavior of the pyrenyl LNA monomer PyL
(Fig. 1) being locked in a C3′-endo RNA-like furanose conformation. The average duplex melting temperatures (Tm values) were decreased by ∼10 °C relative to the native DNA-DNA duplex7,8 which indicated PyL as being more destabilizing than the corresponding pyrenyl DNA monomer for which decreased Tm values of ∼5 °C were reported in a similar sequence context.6 In order to study the relation between conformation and the effect of a pyrenyl unit in relation to universal hybridization, we decided to incorporate the pyrenyl α-L-LNA monomer αLPyL containing a 2′-O,4′-C-methylene-α-L-ribofuranosyl moiety (Fig. 1).10¶ As CD and NMR spectroscopy studies of α-L-LNA:DNA duplexes have shown the overall duplex geometry to be of the B-type with preserved Watson–Crick base pairing, α-L-LNA can be most adequately described as a DNA mimic.11 Therefore, the pyrenyl α-L-LNA monomer αLPyL should furnish information on the effect of the pyrenyl aglycon when locked in a DNA-like conformation, and allow direct comparison with PyL locked in an RNA-like conformation.7,8 In addition, the corresponding phenyl analogue αLPhL
(Fig. 1) was also synthesized.
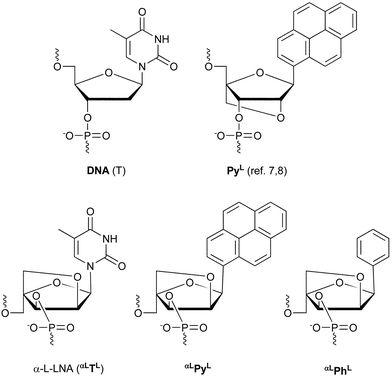 |
| | Fig. 1 Structures of nucleotide monomers: DNA (T), α-L-LNA (αLTL),10 LNA-type pyrenyl monomer (PyL)7,8 and α-L-LNA-type pyrenyl (αLPyL) and phenyl (αLPhL) monomers. The short notations shown are used in Table 1. For DNA and α-L-LNA, the thymine monomers are shown. | |
Table 1 Thermal denaturation experiments (Tm values shown) for ON1–ON6 towards DNA complements with each of the four natural bases in the central positiona
| | Y |
|---|
| DNA Target | 3′-d(CACTYTACG) | A | C | G | T |
|---|
| Melting temperatures (Tm values/°C) measured as the maximum of the first derivative of the melting curve (A260vs. temperature) recorded in medium salt buffer. (10 mM sodium phosphate, 100 mM sodium chloride, 0.1 mM EDTA, pH 7.0) or in high salt buffer. (10 mM sodium phosphate, 700 mM sodium chloride, 0.1 mM EDTA, pH 7.0) using 1.0 µM concentrations of the two strands; A = adenin-9-yl monomer, C = cytosin-1-yl monomer, G = guanin-9-yl monomer, T = thymin-1-yl; see Fig. 1 for structures of αLTL,αLPhL,PyL and αLPyL. |
|---|
| ON1 | 5′-d(GTGATATGC) | 27b/36c | | | |
| ON2 | 5′-d(GTGAPyLATGC)7,8 | 18b | 17b | 18b | 19b |
| ON3 | 5′-d(GTGAαLPhLATGC) | <5b/12c | <5b,c | <5b,c | <5b/12c |
| ON4 | 5′-d(GTGAαLPyLATGC) | 21b/30c | 22b/31c | 27b/34c | 23b/33c |
| ON5 | 5′-d(GαLTLGAαLPhLAαLTLGC) | <5b/14c | <5b,c | <5b,c | <5b/13c |
| ON6 | 5′-d(GαLTLGAαLPyLAαLTLGC) | 24b/33c | 23b/33c | 31b/36c | 26b/34c |
Results and discussion
Synthesis of phosphoramidite building blocks 19a and 19b
As an appropriate synthetic route, the protocol shown in Scheme 1, based on earlier syntheses published for C-aryl nucleosides of LNA,7,8,12–14 was used (yields are shown in the caption of Scheme 1). 1,2:5,6-Di-O-isopropylidene-α-D-glucose was p-methoxybenzylated to give furanoside 1 followed by selective cleavage of the 5,6-O-isopropylidene group with 60% acetic acid at room temperature furnishing diol 2. Periodate cleavage of 2 and subsequent in situ aldol condensation and Cannizzaro reaction with formaldehyde under alkaline conditions afforded furanoside 3. Di-O-mesylation followed by methanolysis afforded a (1 ∶ 2.2) anomeric mixture of methyl furanoside 5 which subsequently was separated by column chromatography. The single anomers were reacted individually in the following steps. Epimerization at C2 to give 2-O-acetyl derivatives 6 was achieved by activation with trifluoromethanesulfonic anhydride followed by nucleophilic substitution by reaction with potassium acetate in the presence of 18-crown-6 ether. Deacetylation with saturated methanolic ammonia yielded mainly furanoside 7 along with traces of the bicyclic compound 8. Analytical samples were obtained after column chromatographic purification, and the crude mixture was directly cyclized in the presence of sodium hydride in DMF to give the 2,5-dioxabicyclo[2.2.1]heptane derivative 8. Substitution of the remaining mesyloxy group of 8 with an acetate group, followed by deacetylation and then p-methoxybenzylation yielded the furanoside 11
(via9 and 10). Due to ring strain, acetal hydrolysis cleanly afforded the monocyclic aldehyde 12 which served as a general electrophile for Grignard additions as reported earlier for derivatives in the enantiomeric series.7,8,12–14 Reaction of this aldehyde with phenyl or pyrenyl Grignard reagents yielded diastereoselectively a single product assigned as the S-epimer based upon the fact that similar Grignard reactions on the enantiomeric aldehyde have been reported to exclusively furnish the R-epimeric products.7,8 This stereochemical assignment was later confirmed (see below) by an NOE experiment on the bicyclic nucleoside 16. For the synthesis of the desired 2′-O,4′-C-methylene-α-L-ribofuranosyl nucleosides another epimerization was required, this time at C1. In situ oxidation of the phenyl analogue 13a with Dess–Martin periodinane (to give ketone 14) followed by reduction with sodium borohydride afforded a 1 ∶ 2.4 separable mixture of diastereomers. Although the epimerized product 15a was isolated in only 22% yield this route was preferred over the alternative, but more complicated, process involving selective 4-O-protection, 1-O-activation, inversion and deprotection. Moreover, the other epimer 13a was recovered. Surprisingly, oxidation of the pyrenyl analogue 13b with the Dess–Martin reagent did not afford the required oxidation product 14b, probably due to the formation of acetic acid during the course of the reaction as it was later observed that 14b is unstable even on silica gel. Instead, the oxidation was carried out under neutral conditions with pyridinium dichromate as oxidant and the crude product obtained was directly reduced without purification to afford a 1 ∶ 2 mixture of epimers 15b and 13b which were separated by column chromatography. The diols 15a and 15b were efficiently cyclized under Mitsunobu conditions to afford the bicyclic α-L-configured C-aryl nucleosides 16a and 16b, respectively. The assigned configuration of compound 16a was verified by an NOE difference experiment. Thus, selective irradiation of one of the H5″ protons (appearing at δ 4.21) gave a 13.2% enhancement of the signal of the pyrenyl moiety and vice versa
(5.8%). In addition, very significant and mutual NOE effects were observed between H1′ and H3′
(11.3/8.7%) which besides verification of configuration strongly suggest that the bicyclic nucleoside adopts the expected N-type (C3′-endo) furanose conformation.¶ Similarly for the bicyclic phenyl derivative 16b mutual NOE enhancements were observed between H1′ and H3′
(10.7/9.1%) and between protons of the phenyl moiety and the H5″ protons (1.4/2.7%). Oxidative removal of the p-methoxybenzyl protection groups was achieved with DDQ yielding derivatives 17a and 17b. To prepare for automated synthesis of oligonucleotides containing the phenyl (αLPhL) and pyrenyl (αLPyL)
α-L-LNA monomers, the primary hydroxy group of nucleoside diols 17a and 17b was selectively protected by reaction with 4,4′-dimethoxytrityl chloride (to give compounds 18a and 18b, respectively) followed by phosphitylation under standard conditions to furnish the desired phosphoramidite building blocks 19a and 19b in satisfactory yields (Scheme 1).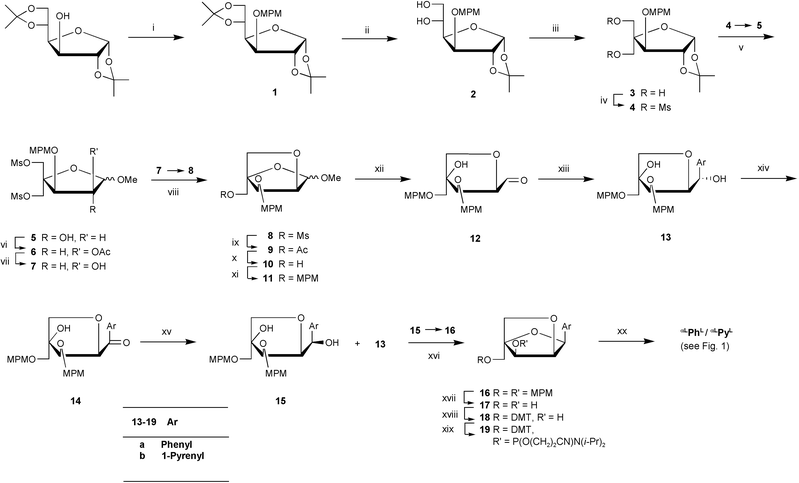 |
| | Scheme 1 Reagents and conditions (and yields): (i)
p-Methoxybenzyl chloride, NaH, THF, (n-Bu)4N+I−
(92%); (ii) 60% AcOH (82%); (iii) (a) NaIO4, H2O, THF, (b) HCHO, 2 M aq. NaOH, dioxane (87%); (iv) MsCl, pyridine (93%); (v) H2O–HCl–CH3OH (1 ∶ 1.5 ∶ 8.5, v/v/v)
(major isomer 5a: 57%
+ minor isomer 5b: 26%); (vi) (a) Tf2O, pyridine, (b) KOAc, 18-crown-6 ether, toluene (6a: 83%; 6b 77%); (vii) sat. methanolic ammonia; (viii) NaH, DMF (8a: 72%
(from 6a); 8b: 69%
(from 6b); (ix) KOAc, 18-crown-6 ether, dioxane (9a: 92%; 9b: 78%); (x) sat. methanolic ammonia (10a: 96%; 10b: 90%); (xi)
p-methoxybenzyl chloride, NaH, THF, (n-Bu)4N+I−
(11a: 86%; 11b: 81%); (xii) 70% AcOH (85% from 11a; 80% from 11b); (xiii) ArMgBr, THF (13a: 74%; 13b: 71%); (xiv) Dess–Martin periodinane, CH2Cl2
(14a: 76%); PDC, 3Å molecular sieves, CH2Cl2
(14b); (xv) NaBH4, THF, H2O (15a: 22%; 15b:16%
(from 13b)); (xvi) TMAD, Bu3P, benzene (16a: 97%; 16b: 88%); (xvii) DDQ, CH2Cl2, H2O (17a: 63%; 17b: 52%); (xviii) DMTCl, pyridine (18a: 88%; 18b: 54%); (xix) NC(CH2)2OP(Cl)N(i-Pr)2, EtN(i-Pr)2, CH2Cl2
(19a: 57%; 19b: 42%); (xx) DNA synthesizer. | |
Synthesis of oligonucleotides containing C-phenyl monomer αLPhL and C-pyrenyl monomer αLPyL
All oligonucleotides ON3–ON6
(Table 1) were prepared in 0.2 µmol scale using the phosphoramidite approach (see the Experimental section for details). The stepwise coupling efficiencies of phosphoramidites 19a and 19b
(10 min coupling time) and of unmodified deoxynucleoside phosphoramidites (2 min coupling time) were >99% using 1H-tetrazole as activator (pyridine hydrochloride was used as activator when coupling 19b). After standard deprotection and cleavage from the solid support using 32% aqueous ammonia (12 h, 55 °C), the oligomers were purified by precipitation from ethanol. The composition of oligomers ON3–ON6 was verified by MALDI-MS analysis and their purity (>80%) by capillary gel electrophoresis.Thermal denaturation studies and fluorescence measurements
The hybridization of the novel modified oligonucleotides ON3–ON6
(Table 1) towards four 9-mer DNA targets with the central base being each of four natural bases was studied by thermal denaturation experiments at medium and high salt buffer conditions (see the Experimental section for details). The obtained melting temperatures (Tm values) were compared with those obtained for the DNA reference ON1
(towards the fully matched target) and with those reported for the pyrenyl LNA monomer PyL showing universal hybridization.7,8 Introduction of the phenyl α-L-LNA monomer αLPhL
(ON3) reduces the thermal stability of the resulting duplexes very significantly even at high salt conditions and even when combined with two thymine α-L-LNA monomers (monomers αLTL, ON5) known to be affinity enhancing in this sequence context.15 Interestingly, incorporation of one pyrenyl α-L-LNA monomer αLPyL
(ON4) induces Tm values in both medium and high salt conditions being only moderately reduced relative to, or similar to, the DNA reference ON1. In this case, the introduction of two thymine α-L-LNA monomers (ON6) has a weak positive effect on the Tm values. It is apparent from the studies on ON4 and ON6 that the pyrenyl α-L-LNA monomer αLPyL displays a subtle preference for “pairing” with a guanine monomer, and that this effect is most pronounced under medium salt conditions.Comparison of the pyrenyl LNA monomer PyL
(ON2)7,8 and the pyrenyl α-L-LNA monomer αLPyL
(ON4) reveals several interesting differences. Under medium salt conditions, the affinity was determined7,8 to be significantly lower for PyL but the hybridization with the four DNA target strands more universal. However, the performance of the pyrenyl α-L-LNA monomer αLPyL as a universal base is improved by changing into high salt conditions (Tm values = 30–34 °C (ON4) and 33–36 °C (ON6).
Fluorescence spectra of single stranded ON4 and of duplexes formed between ON4 and the four 9-mer DNA targets were measured (Fig. 2). In general, the spectra show the characteristics of pyrene monomer emission, i.e., two bands (band I and III) with emission maxima at 383 and 401 nm, respectively. The fluorescence intensity of single stranded ON4 is about 45% of that of the monomer 17b, and stronger than the intensities obtained for the four duplexes (Fig. 2). These facts support intercalation of the pyrene aglycon of αLPyL, especially in the case of the duplexes resulting in more efficient fluorescence quenching by the nucleobases than for the more flexible single stranded ON4. It is noticeable that the minimum fluorescence intensity is obtained for the thermally most stable duplex which underlines the importance of intercalation and/or reduced flexibility for fluorescence quenching.
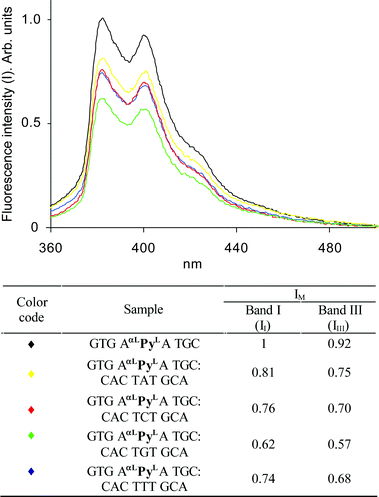 |
| | Fig. 2 Steady state fluorescence spectra of single stranded ON4 and of the four duplexes shown. The spectra were recorded at 19 °C ± 1 °C in buffer (10 mM sodium phosphate, 700 mM sodium chloride, 0.1 mM EDTA, pH 7.0) using 1.5 × 10−7 M concentrations of each strand. Fluorescence intensities are arbitrary units normalized relative to the fluorescence intensity of band I of ON4
(assigned the value “1”). | |
Anticipating intercalation, at least in part, of the pyrenyl moiety of αLPyL into the nucleobase stack, the results obtained herein show pyrenyl intercalation and nucleobase interaction to be more efficient in the DNA-like context of ON4 than in the RNA-like context of the corresponding LNA (PyL).7,8 A similar trend has been reported for other pyrene-containing monomers.16–19 However, comparison of relative melting temperatures obtained with the pyrenyl α-L-LNA monomer αLPyL and the corresponding pyrenyl DNA monomer6 reveals no affinity enhancing effect of conformational restriction. It is plausible that the limited flexibility imposed by the locked 2,5-dioxabicyclo[2.2.1]heptane skeleton impedes optimal intercalation of the pyrenyl moiety.
Conclusion
Synthesis of phenyl and pyrenyl derivatives of α-L-LNA-type C-aryl nucleosides has been accomplished as has their efficient incorporation into 9-mer DNA and α-L-LNA strands. For the pyrenyl derivative, universal hybridization towards DNA targets was obtained under high salt conditions ([Na+]
= 710 mM). Fluorescence measurements indicated intercalation of the pyrenyl moiety into the nucleobase stack of the duplexes.Experimental
General
Reactions were conducted under an atmosphere of nitrogen when anhydrous solvents were used. All reactions were monitored by thin-layer chromatography (TLC) using silica gel coated plates with fluorescence indicator (SiO2-60, F-254) which were visualized under UV light and by spraying with 5% conc. sulfuric acid in absolute ethanol (v/v) followed by heating. Silica gel 60 (particle size 0.040–0.063 mm, Merck) was used for column chromatography. Light petroleum of the distillation range 60–80 °C was used. After column chromatography, fractions containing product were pooled, evaporated under reduced pressure and dried 12 h under high vacuum to give the product unless otherwise specified. 1H NMR spectra were recorded at 300 MHz, 13C NMR spectra at 75.5 MHz, and 31P NMR spectra at 121.5 MHz. Chemical shifts are reported in ppm relative to either tetramethylsilane or the deuterated solvent as internal standard for 1H NMR and 13C NMR, and relative to 85% H3PO4 as external standard for 31P NMR. Assignments of NMR spectra, when given, are based on 2D NMR experiments (the assignments of methylene protons/carbons may be interchanged). Coupling constants (J values) are given in Hertz. Bicyclic compounds are named according to the Von Bayer nomenclature, whereas the atom numbering follows the standard carbohydrate/nucleoside nomenclature. Fast atom bombardment mass spectra (FAB-MS) were recorded in positive ion mode on a Kratos MS50TC spectrometer and MALDI-HRMS were recorded in positive ion mode on a IonSpec Fourier transform mass spectrometer. The composition of the oligonucleotides was verified by MALDI-MS on a Micromass Tof Spec E mass spectrometer using a matrix of diammonium citrate and 2,6-dihydroxyacetophenone.3-O-(p-Methoxybenzyl)-1,2:5,6-di-O-isopropylidene-α-D-glucofuranose (1). Sodium hydride (4.5 g, 60% suspension in mineral oil, 112.5 mmol) was added to anhydrous THF (30 cm3) under an inert atmosphere and the suspension was cooled to 0 °C. A solution of 1,2:5,6-di-O-isopropylidene-α-D-glucofuranose (19.5 g, 74.9 mmol) in anhydrous THF (20 cm3) was added dropwise over 30 min. The temperature of the mixture was allowed to raise to rt and stirring was continued for 15 min. p-Methoxybenzyl chloride (14.5 g, 92.6 mmol) was added dropwise followed by tetrabutylammonium iodide (8.6 g, 23.3 mmol) and the contents were stirred for 36 h at rt. The mixture was cooled to 0 °C and ice-cold water (100 cm3) was carefully added. Extraction was performed with EtOAc (2 × 100 cm3) and the combined organic phase was washed with brine (60 cm3), dried (Na2SO4), filtered and evaporated to dryness under reduced pressure. The residue was purified by column chromatography [8–10%
(v/v) EtOAc in light petroleum] to give furanoside 1 as a colourless oil (26.2 g, 92%). Rf 0.33 (EtOAc–light petroleum 75 ∶ 25, v/v); δH
(CDCl3) 7.27 (2H, d, J 8.7, Ar-H), 6.87 (2H, d, J 8.6, Ar-H), 5.88 (1H, d, J 3.8, H-1), 4.63–4.54 (3H, m, H-2 and CH2
(MPM)), 4.33 (1H, m, H-4), 4.16–4.08 (2H, m, H-3 and H-6a), 4.02–3.97 (2H, m, H-5 and H-6b), 3.80 (3H, s, OCH3), 1.49, 1.43, 1.38, 1.31 (3H each, 4s, CH3
(isopropylidene)); δC
(CDCl3) 159.5, 129.8, 129.4 and 113.9 (ArH), 111.8 and 109.0 (OC(CH3)2O), 105.4 (C-1), 82.8 (C-2), 81.4 (C-3 and C-5), 72.7 (C-4), 72.2 (CH2
(MPM)), 67.5 (C-6), 55.4 (OCH3), 26.9, 26.8, 26.3 and 25.6 (CH3
(isopropylidene)), MALDI-HRMS: m/z 403.1714 ([M + Na]+, C20H28O7Na+ calc. 403.1727).
1,2-Di-O-isopropylidene-3-O-(p-methoxybenzyl)-α-D-glucofuranose (2). 60% Acetic acid (300 cm3) was added to furanoside 1
(26.0 g, 68.3 mmol) and the reaction mixture was stirred at rt for 24 h. The reaction mixture was washed with light petroleum (2 × 100 cm3) and subsequently concentrated to dryness under reduced pressure and co-evaporated successively with absolute ethanol (2 × 50 cm3) and toluene (2 × 50 cm3). The residue was dissolved in CH2Cl2
(200 cm3) and washed with sat. aq. NaHCO3
(2 × 100 cm3). The organic phase was dried (Na2SO4), filtered and evaporated to dryness under reduced pressure to give a residue which was purified by column chromatography [40%
(v/v) EtOAc in light petroleum] to give furanoside 2 as a colourless oil (19.1 g, 82%). Rf 0.28 (EtOAc–light petroleum 50 ∶ 50, v/v); δH
(CDCl3) 7.27 (2H, d, J 8.7, Ar-H), 6.89 (2H, d, J 8.3, Ar-H), 5.92 (1H, d, J 3.6, H-1), 4.66 (1H, d, J 11.6, CH2
(MPM)), 4.60 (1H, d, J 3.9, H-2), 4.47 (1H, d, J 11.6, CH2
(MPM)), 4.13–4.07 (2H, m, H-3 and H-4), 4.00 (1H, m, H-5), 3.79 (3H, s, OCH3), 3.77 (1H, m, H-6a), 3.67 (1H, m, H-6b), 2.68 (1H, d, J 5.7, 5-OH), 2.44 (1H, br s, 6-OH), 1.48 and 1.31 (3H each, 2s, CH3
(isopropylidene)); δC
(CDCl3) 159.7, 129.7, 129.3 and 114.2 (Ar), 111.9, (OC(CH3)2O), 105.2 (C-1), 82.2 (C-2), 81.6 and 80.0 (C-3 and C-4), 71.8 (CH2
(MPM)), 69.4 (C-5), 64.4 (C-6), 55.4 (OCH3), 26.8 and 26.3 (CH3
(isopropylidene)); MALDI-HRMS: m/z 363.1400 ([M + Na]+, C17H24O7Na+ calc. 363.1414).
1,2-O-Isopropylidene-4-C-hydroxymethyl-3-O-(p-methoxybenzyl)-β-L-threo-pentofuranose (3). Sodium periodate (38 g, 177.7 mmol) was added slowly to a stirred solution of furanoside 2
(50.0 g, 146.9 mmol) in a (1 ∶ 1) mixture of THF-H2O (360 cm3) at 0 °C. The reaction mixture was stirred 12 h at rt. The white precipitate formed was filtered off and washed with EtOAc (3 × 50 cm3). The aqueous phase was separated and extracted with EtOAc (2 × 60 cm3), and the combined organic phase was washed with brine (2 × 50 cm3) and evaporated to dryness under reduced pressure. The viscous oil obtained was dissolved in dioxane (130 cm3) and formaldehyde (37% solution in water, 34.4 cm3, 391.4 mmol) and sodium hydoxide (2 M solution, 150 cm3) were added. The reaction mixture was stirred for 24 h at rt whereupon extraction was performed with CH2Cl2
(2 × 200 cm3). The combined organic phase was washed with brine (150 cm3), dried (Na2SO4), filtered and evaporated to dryness under reduced pressure. The residue was purified by column chromatography [50%
(v/v) EtOAc in light petroleum] to give furanoside 3 as a white solid material (43.3 g, 87%).Rf 0.28 (EtOAc–light petroleum 50 ∶ 50, v/v); δH
(CDCl3) 7.25 (2H, d, J 8.6, Ar-H), 6.89 (2H, d, J 8.5, Ar-H), 6.00 (1H, d, J 4.4, H-1), 4.74 (dd, 1H, J 1.5 and 4.5, H-2), 4.69 (1H, d, J 11.3, CH2
(MPM)), 4.47 (1H, d, J 11.3, CH2
(MPM)), 4.09 (1H, d, J 1.9, H-3), 3.81 (3H, s, OCH3), 3.72–3.60 (4H, m, H-5 and H-5′), 2.44–2.41 (2H, m, 2 × OH), 1.54 and 1.35 (3H each, 2 s, CH3
(isopropylidene)); δC
(CDCl3) 159.7, 129.6, 129.0 and 114.1 (Ar), 113.2 (OC(CH3)2O), 105.0 (C-1), 89.9 (C-4), 86.0 (C-2), 84.6 (C-3), 72.4 (CH2
(MPM)), 63.9 and 63.5 (C-5 and C-5′), 55.4 (OCH3), 27.4 and 26.9 (CH3
(isopropylidene)); MALDI-HRMS: m/z 363.1402 ([M + Na]+, C17H24O7Na+ calc. 363.1414).
1,2-O-Isopropylidene-5-O-methanesulfonyl-4-C-methanesulfonyloxymethyl-3-O-(p-methoxybenzyl)-β-L-threo-pentofuranose (4). Methanesulfonyl chloride (6.39 g, 55.8 mmol) was added dropwise to a stirred solution of furanoside 3
(7.60 g, 22.3 mmol) in a mixture of anhydrous pyridine (10 cm3) and CH2Cl2
(10 cm3). The reaction mixture was stirred 12 h at rt. The mixture was evaporated to dryness under reduced pressure and the residue was dissolved in CH2Cl2
(50 cm3) and washed with sat. aq. NaHCO3
(2 × 25 cm3). The organic phase was dried (Na2SO4), filtered and concentrated to dryness under reduced pressure. The residue obtained was purified by column chromatography [50%
(v/v) EtOAc in light petroleum] affording furanose 4 which was crystallized from EtOH as a white solid material (10.3 g, 93%). Rf 0.45 (EtOAc–light petroleum 50 ∶ 50, v/v); δH
(CDCl3) 7.26.(2H, d, J 8.5, Ar-H), 6.90 (2H, d, J 8.8, Ar-H), 5.97 (1H, d, J 3.9, H-1), 4.69 (1H, d, J 3.9, H-2), 4.64 (1H, d, J 11.3, CH2
(MPM)), 4.48 (1H, d, J 11.2, CH2
(MPM)), 4.39–4.30 (4H, m, H-5 and H-5′), 4.07 (1H, s, H-3), 3.81 (3H, s, OCH3), 3.03 (3H, s, SO2CH3), 3.00 (3H, s, SO2CH3), 1.57 and 1.32 (3H each, 2s, (CH3(isopropylidene)); δC
(CDCl3) 159.8, 129.9, 128.4 and 114.1 (Ar), 113.3 (OC(CH3)2O), 105.9 (C-1), 86.1 (C-4), 84.3 (C-2), 82.5 (C-3), 72.7 (CH2
(MPM)), 67.5 and 67.3 (C-5 and C-5′), 55.4 (OCH3), 37.7 and 37.5 (2 × SO2CH3), 26.6 and 26.0 (CH3
(isopropylidene)); MALDI-HRMS: m/z 519.0965 ([M + Na]+, C19H28O11S2Na+ calc. 519.0965).
Methyl 5-O-methanesulfonyl-4-C-methanesulfonyloxymethyl-3-O-(p-methoxybenzyl)-α,β-L-threo-pentofuranoside (5). A suspension of furanoside 4
(10.5 g, 21.2 mmol) in a mixture of concentrated aqueous HCl and MeOH (380 cm3, 3 : 17, v/v) was stirred at rt for 36 h. The mixture was carefully neutralized by addition of NaHCO3
(s), whereupon the mixture was evaporated to dryness under reduced pressure. H2O (100 cm3) was added to the residue obtained and extraction was performed with EtOAc (3 × 100 cm3). The combined organic phase was washed with brine (100 cm3), dried (Na2SO4), filtered and concentrated to dryness under reduced pressure. The residue was coevaporated with toluene (2 × 25 cm3) and purified by column chromatography [45–50%
(v/v) EtOAc in light petroleum] to give first the minor isomer 5b
(2.6 g, 26%) and then [50–60%
(v/v) EtOAc in light petroleum] the major isomer 5a
(5.7 g, 57%) both as a clear oil. Rf 0.52, 0.43 (EtOAc–light petroleum 75 ∶ 25, v/v); δH
(CDCl3, major isomer 5a) 7.27 (2H, d, J 8.8, Ar-H), 6.89 (2H, d, J 8.8, Ar-H), 4.89 (1H, d, J 4.8, H-1), 4.74 (1H, d, J 11.5, CH2
(MPM)), 4.59 (1H, d, J 11.5, CH2
(MPM)), 4.35 (1H, d, J 11.0, H-5a), 4.31 (1H, m, H-2), 4.27 (1H, d,J 10.9, H-5b), 4.24 (1H, d, J 10.2, H-5′a), 4.06 (1H, d, J 10.1, H-5′b), 3.98 (1H, d, J 7.0, H-3), 3.80 (3H, s, OCH3
(MPM)), 3.46 (3H, s, OCH3), 3.02 and 2.96 (3H each, 2s, SO2CH3), 2.54 (1H, d, J 9.7, 2-OH); δC
(CDCl3, major isomer) 159.6, 129.8, 129.2 and 114.0 (Ar), 101.8 (C-1), 84.6 (C-3), 80.9 (C-4), 77.5 (C-2), 72.6 (CH2
(MPM)), 69.5 and 68.7 (C-5 and C-5′), 56.1 (OCH3), 55.3 (OCH3
(MPM)), 37.6 and 37.5 (2 × SO2CH3); MALDI-HRMS: m/z 493.0815 ([M + Na]+, C17H26O11S2Na+ calc. 493.0809); δH
(CDCl3, minor isomer 5b) 7.27 (2H, d, J 8.5, Ar-H), 6.90 (2H, d, J 8.6, Ar-H), 4.87 (1H, s, H-1), 4.63 (1H, d, J 11.7, H-5a), 4.51 (1H, d, J 11.7, H-5b), 4.44 (1H, d, J 10.7, H-5′a), 4.35 (1H, d,J 10.6, H-5′b), 4.26 (3H, m, H-2 and CH2
(MPM)), 3.97 (1H, d, J 1.9, H-3), 3.81 (3H, s, OCH3
(MPM)), 3.41 (3H, s, OCH3), 3.01 and 3.00 (3H each, 2s, 2 × SO2CH3), 2.47 (1H, d, J 4.3, 2-OH); δC
(CDCl3, minor isomer) 159.7, 129.9, 129.1, 114.1, 110.2, 85.3, 84.3, 79.9, 72.9, 69.1, 68.9, 55.9, 55.4, 37.6 and 37.3; MALDI-HRMS: m/z 493.0817 ([M + Na]+, C17H26O11S2Na+ calc. 493.0809).
Methyl 2-O-acetyl-5-O-methanesulfonyl-4-C-methanesulfonyloxymethyl-3-O-(p-methoxybenzyl)-α,β-L-erythro-pentofuranoside (6). To a stirred solution of furanoside 5a
(18.1 g, 38.5 mmol) in pyridine (38 cm3) and CH2Cl2
(176 cm3) was dropwise added triflic anhydride (13.0 g, 46.1 mmol) at −30 °C over 30 min. The resulting mixture was stirred for 12 h at rt and then washed with sat. aq. NaHCO3
(2 × 100 cm3), dried (Na2SO4), filtered and evaporated to dryness under reduced pressure. The residue was coevaporated with toluene (2 × 25 cm3) and then dissolved in a mixture of CH2Cl2
(53 cm3) and toluene (400 cm3). 18-crown-6 ether (32 g, 121.0 mmol) and KOAc (12.8 g, 130.4 mmol) were added to the stirred solution. After 12 h at 50 °C, the reaction mixture was washed with sat. aq. NaHCO3
(2 × 100 cm3), dried (Na2SO4), filtered and concentrated to dryness under reduced pressure. The residue was purified by column chromatography [30–35%
(v/v) EtOAc in light petroleum] to give furanoside 6a as a white solid material (16.4 g, 83%). Rf 0.69 (EtOAc–light petroleum 75 ∶ 25, v/v); δH
(CDCl3) 7.23 (2H, d, J 8.3, Ar-H), 6.88 (2H, d, J 8.7, Ar-H), 5.27 (1H, d, J 4.8, H-2), 4.90 (1H, s, H-1), 4.52 (1H, d, J 10.8, CH2
(MPM)), 4.46–4.31 (5H, m, H-3, H-5, H-5′a and CH2
(MPM)), 4.09 (1H, d, J 9.9, H-5′b), 3.80 (3H, s, OCH3
(MPM)), 3.36 (3H, s, OCH3), 3.03 and 3.01 (3H each, 2s, 2 × SO2CH3), 2.13 (3H, s, COCH3); δC
(CDCl3) 169.6 (COCH3), 159.8, 130.0, 128.8 and 114.0 (Ar), 105.9 (C-1), 81.3 (C-4), 79.9 (C-3), 74.0 (C-2), 73.5 (CH2
(MPM)), 69.3 and 69.1 (C-5 and C-5′), 55.6 (OCH3), 55.4 (OCH3
(MPM)), 37.6 and 37.5 (2 × SO2CH3), 20.8 (COCH3); MALDI-HRMS: m/z 535.0913 ([M + Na]+, C19H28O12S2Na+ calc. 535.0914); Similarly the minor isomer 5b
(9.0 g, 19.2 mmol) was activated with triflic anhydride (7.0 g, 24.8 mmol) in the presence of pyridine (20 cm3), followed by epimerization with KOAc (6.42 g, 65.4 mmol) in the presence of 18-crown-6 ether (17.6 g, 66.6 mmol). The residue obtained after work up procedure was purified by column chromatography [40–50%
(v/v), EtOAc in light petroleum] to give furanoside 6b as a white solid material (7.55 g, 77%). Rf 0.58 (EtOAc–light petroleum 75 ∶ 25, v/v); δH
(CDCl3) 7.27 (2H, d, J 8.3, Ar-H), 6.89 (2H, d, J 8.0, Ar-H), 5.12 (1H, d, J 4.4, H-1), 4.95 (1H, dd, J 5.0 and 6.3, H-2), 4.65 (1H, d, J 11.4, H-5a), 4.50 (1H, d, J 11.1, H-5′a), 4.43 (1H, d, J 11.7, H-5b), 4.24 (1H, d, J 6.8, H-3), 4.19 (1H, J 10.8, H-5′b), 4.12 (2H, s, CH2
(MPM)), 3.81 (3H, s, OCH3
(MPM)), 3.46 (3H, s, OCH3), 3.00 and 2.99 (3H each, 2s, 2 × SO2CH3), 2.16 (3H, s, COCH3); δC
(CDCl3) 170.2 (COCH3), 159.9, 130.3, 128.8 and 114.1 (Ar), 102.7 (C-1), 82.7 (C-4), 76.5 (C-3), 74.6 (CH2
(MPM)), 71.7 (C-2), 69.3 and 68.8 (C-5 and C-5′), 56.5 (OCH3), 55.4 (OCH3
(MPM)), 37.6 and 37.3 (SO2CH3), 20.8 (COCH3); MALDI-HRMS: m/z 535.0927 ([M + Na]+, C19H28O12S2Na+ calc. 535.0914).
Methyl 5-O-methanesulfonyl-4-C-methanesulfonyloxymethyl-3-O-(p-methoxybenzyl)-α,β-L-erythro-pentofuranoside (7). A solution of furanoside 6a
(isomer obtained from the major anomer of 5, 22.0 g, 42.9 mmol) in sat. methanolic ammonia (250 cm3) was stirred for 24 h at rt (complete conversion of starting material into two products according to analytical TLC). The reaction mixture was evaporated to dryness under reduced pressure and the residue was coevoporated with toluene (2 × 50 cm3). An analytical sample was purified by column chromatography (30%
(v/v) EtOAc in light petroleum) to give as the major product furanoside 7a. Rf 0.53 (EtOAc–light petroleum 75 ∶ 25, v/v); δH
(CDCl3) 7.28 (2H, d, J 8.3, Ar-H), 6.91 (2H, d, J 8.6, Ar-H), 4.87 (1H, s, H-1), 4.62 (1H, d, J 11.4, CH2
(MPM)), 4.53 (1H, d, J 11.2, CH2
(MPM)), 4.43 (1H, d, J 11.1, H-5a), 4.38 (1H, d, J 11.3, H-5b), 4.31 (1H, d, J 9.6, H-5′a), 4.25 (1H, d, J 4.9, H-3), 4.06 (1H, d, J 9.8, H-5′b), 3.98 (1H, dd, J 2.5 and 4.7, H-2), 3.81 (3H, s, OCH3
(MPM)), 3.33 (3H, s, OCH3), 3.06 (3H, s, SO2CH3), 3.03 (3H, s, SO2CH3), 2.55 (1H, d, J 2.5, 2-OH); δC
(CDCl3) 160.0, 130.1, 128.5 and 114.3 (Ar), 107.8 (C-1), 81.7 (C-4), 81.2 (C-3), 73.9 (C-2), 73.6 (CH2
(MPM)), 69.7 and 69.5 (C-5 and C-5′), 55.5 (OCH3), 55.4 (OCH3
(MPM)), 37.5 (SO2CH3), 37.4 (SO2CH3); MALDI-HRMS: m/z 493.0811 ([M + Na]+, C17H26O11S2Na+ calc. 493.0808). The minor product was tentatively assigned as 8a presumably formed by deacetylation followed by base induced intramolecular cyclization. The crude mixture of 7a and 8a was therefore used in the next step without purification. Similarly, deacetylation of the epimer 6b
(5.05 g, 9.85 mmol) using sat. methanolic ammonia (60 cm3) yielded two products. An analytical sample was purified by column chromatography (35%
(v/v) EtOAc in light petroleum) to give the major product 7b as a white solid material. Rf 0.46 (EtOAc–light petroleum 75 ∶ 25, v/v); δH
(CDCl3) 7.28 (2H, d, J 8.5, Ar-H), 6.91 (2H, d, J 8.5, Ar-H), 4.88 (1H, d, J 4.7, H-1), 4.70 (1H, d, J 11.3, CH2
(MPM)), 4.52 (1H, d, J 11.4, CH2
(MPM)), 4.44 (1H, d, J 10.8, H-5a), 4.20 (1H, d, J 11.0, H-5b), 4.18 (1H, m, H-2), 4.14 (1H, d, J 10.5, H-5′a), 4.08 (1H, d, J 10.6, H-5′b), 4.00 (1H, d, J 7.0, H-3), 3.81 (3H, s, OCH3
(MPM)), 3.46 (3H, s, OCH3), 3.01 (3H, s, SO2CH3), 3.00 (3H, s, SO2CH3), 2.88 (1H, d, J 11.2, 2-OH); δC
(CDCl3) 159.8, 130.2, 128.9 and 114.2 (Ar), 103.6 (C-1), 83.2 (C-4), 77.7 (C-3), 75.0 (C-2), 71.9 (CH2
(MPM)), 69.2 and 69.1 (C-5 and C-5′), 56.3 (OCH3), 55.4 (OCH3
(MPM)), 37.6 (SO2CH3), 37.4 (SO2CH3); MALDI-HRMS: m/z 493.0819 ([M + Na]+, C17H26O11S2Na+ calc. 493.0809).
(1S,3RS,4S,7R)-1-Methanesulfonyloxymethyl-3-methoxy-7-(p-methoxybenzyloxy)-2,5-dioxabicyclo[2.2.1]heptane (8). The crude mixture (∼23.0 g) obtained from methanolic ammonia treatment of 6a was dissolved in anhydrous DMF (50 cm3) and the resulting mixture was cooled to 0 °C. Sodium hydride (4.0 g, 60% suspension in mineral oil (w/w), 100 mmol) was added slowly during 10 min and the mixture was stirred for 12 h at rt whereupon ice-cold H2O (200 cm3) was carefully added. The resulting mixture was extracted using EtOAc (3 × 100 cm3) and the combined organic phase was washed successively with sat. aq. NaHCO3
(2 × 100 cm3) and brine (50 cm3), dried (Na2SO4), filtered and concentrated to dryness under reduced pressure. The residue was purified by column chromatography (30%
(v/v) EtOAc in light petroleum) to give furanoside 8a as a white solid material (11.6 g, 72% from 6a). Rf 0.35 (EtOAc–light petroleum 50 ∶ 50, v/v); δH
(CDCl3) 7.26 (2H, d, J 8.6, Ar-H), 6.89 (2H, d, J 8.7, Ar-H), 4.81 (1H, s, H-1), 4.59 (1H, d, J 11.2, H-5a) 4.53–4.44 (3H, m, H-5b and CH2
(MPM)), 4.10 and 4.09 (1H each, 2s, H-2 and H-3), 4.00 (1H, d, J 7.4, H-5′a), 3.81 (3H, s, OCH3
(MPM)), 3.69 (1H, d, J 7.4, H-5′b), 3.37 (3H, s, OCH3), 3.05 (3H, s, SO2CH3); δC
(CDCl3) 159.6, 129.5, 129.3 and 114.0 (Ar), 105.3 (C-1), 83.2 (C-4), 78.6 and 77.2 (C-2 and C-3), 72.1 (CH2
(MPM)), 71.8(C-5′), 66.3(C-5), 55.6 (OCH3), 55.4 (OCH3
(MPM)), 37.8 (SO2CH3); MALDI-HRMS: m/z 397.0928 ([M + Na]+, C16H22O8SNa+ calc. 397.0927). Similarly the crude mixture (∼5.5 g) obtained from methanolic ammonia treatment of 6b was reacted with sodium hydride (1.0 g, 60% suspension in mineral oil (w/w), 25 mmol)). After work-up, the residue was purified by column chromatography (45%
(v/v) EtOAc in light petroleum) affording furanoside 8b as a white solid material (2.54 g, 69% from 6b). Rf 0.24 (EtOAc–light petroleum 50 ∶ 50, v/v); δH
(CDCl3) 7.27 (2H, d, J 8.6, Ar-H), 6.89 (2H, d, J 9.0, Ar-H), 4.99 (1H, s, H-1), 4.61 (1H, d, J 11.7, CH2
(MPM)), 4.52 (1H, d, J 11.4, CH2
(MPM)), 4.48 (1H, d, J 11.9, H-5a), 4.41 (1H, d, J 11.6, H-5b), 4.18 (1H, s, H-2), 4.04 (1H, d, J 8.0, H-5′a), 3.92 (1H, d, J 8.8, H-5′b), 3.91 (1H, s, H-3), 3.81 (3H, s, OCH3
(MPM)), 3.47 (3H, s, OCH3), 3.05 (3H, s, SO2CH3); δC
(CDCl3) 159.5, 129.4, 128.8 and 113.9 (Ar), 104.2 (C-1), 86.2 (C-4), 79.2 (C-3), 76.9 (C-2), 72.1 (C-5′), 71.7 (CH2
(MPM)), 66.0 (C-5), 56.2 (OCH3), 55.2 (OCH3
(MPM)), 37.5 (SO2CH3); MALDI-HRMS: m/z 397.0926 ([M + Na]+ calc. C16H22O8SNa+ 397.0927).
(1S,3RS,4S,7R)-1-Acetoxymethyl-3-methoxy-7-(p-methoxybenzyloxy)-2,5-dioxabicyclo[2.2.1]heptane (9). To a stirred solution of furanoside 8a
(11.6 g, 31.0 mmol) in anhydrous dioxane (45 cm3) was added 18-crown-6 (16.4 g, 62.0 mmol) and KOAc (15.2 g, 154.9 mmol), and the mixture was heated under reflux for 12 h. The reaction mixture was concentrated to dryness under reduced pressure and the residue dissolved in CH2Cl2
(100 cm3). Washing was performed successively with sat. aq. NaHCO3
(2 × 50 cm3) and brine (50 cm3). The separated organic phase was dried (Na2SO4), filtered and evaporated to dryness under reduced pressure. The residue was purified by column chromatography (20%
(v/v) EtOAc in light petroleum) to afford furanoside 9a as a white solid material (9.65 g, 92%). Rf 0.63 (EtOAc–light petroleum 50 ∶ 50, v/v); δH
(CDCl3) 7.26 (2H, d, J 7.9, Ar-H), 6.88 (2H, d, J 8.4, Ar-H), 4.80 (1H, s, H-1), 4.59 (1H, d, J 11.6, CH2
(MPM)), 4.48 (1H, d, J 11.7, CH2
(MPM)), 4.47 (1H, d, J 12.7, H-5a), 4.27 (1H, d, J 12.4, H-5b), 4.09 (1H, s, H-2), 4.05 (1H, s, H-3), 3.99 (1H, d, J 7.4, H-5′a), 3.80 (3H, s, OCH3
(MPM)), 3.71 (1H, d, J 7.4, H-5′b), 3.36 (3H, s, OCH3), 2.06 (3H, s, COCH3); δC
(CDCl3) 170.4 (COCH3), 159.3, 129.4, 129.2 and 113.7 (Ar), 104.9 (C-1), 83.1 (C-4), 78.7 (C-2), 76.9 (C-3), 71.9 (CH2
(MPM)), 71.8 (C-5′), 60.9 (C-5), 55.2 and 55.1 (OCH3 and OCH3
(MPM)), 20.6 (COCH3); MALDI-HRMS: m/z 361.1245 ([M + Na]+, C17H22O7Na+ calc. 361.1258). Similarly, reaction of furanoside 8b
(646 mg, 1.73 mmol) with KOAc (880 mg, 8.97 mmol) in the presence of 18-crown-6 ether (940 mg, 3.56 mmol) in anhydrous dioxane (3 cm3) followed by work-up and column chromatography (30%
(v/v) EtOAc–light petroleum) afforded furanoside 9b as a white solid material (455 mg, 78%). Rf 0.29 (EtOAc–light petroleum 50 ∶ 50, v/v); δH
(CDCl3) 7.26 (2H, d, J 9.0, Ar-H), 6.88 (2H, d, J 8.3, Ar-H), 4.98 (1H, s, H-1), 4.61 (1H, d, J 11.7, CH2
(MPM)), 4.49 (1H, d, J 11.7, CH2
(MPM)), 4.35 (1H, d, J 12.9, H-5a), 4.28 (1H, d, J 12.8, H-5b), 4.18 (1H, s, H-2), 4.02 (1H, d, J 8.0, H-5′a), 3.92 (1H, d, J 7.9, H-5′b), 3.87 (1H, s, H-3), 3.80 (3H, s, OCH3
(MPM)), 3.48 (3H, s, OCH3), 2.05 (3H, s, COCH3); δC
(CDCl3) 170.6 (COCH3), 159.6, 129.5, 129.3 and 113.9 (Ar), 104.3 (C-1), 86.6 (C-4), 79.5 (C-3), 77.1 (C-2), 72.6 (C-5′), 71.7 (CH2
(MPM)), 60.8 (C-5), 56.4 (OCH3), 55.4 (OCH3
(MPM)), 20.8 (COCH3); MALDI-HRMS: m/z 361.1253 ([M + Na]+, C17H22O7Na+ calc. 361.1258).
(1R,3RS,4S,7R)-1-Hydroxymethyl-3-methoxy-7-(p-methoxybenzyloxy)-2,5-dioxabicyclo[2.2.1]heptane (10). A solution of furanoside 9a
(2.27 g, 6.71 mmol) in sat. methanolic ammonia (55 cm3) was stirred for 12 h at rt. The reaction mixture was evaporated to dryness under reduced pressure and coevaporated with toluene (2 × 10 cm3). The residue was purified by column chromatography (55%
(v/v) EtOAc in light petroleum) to give furanoside 10a as a white solid material (1.91 g, 96%). Rf 0.31 (EtOAc–light petroleum 50 ∶ 50, v/v); δH
(CDCl3) 7.28–7.25 (2H, m, Ar-H), 6.89–6.86 (2H, m, Ar-H), 4.80 (1H, s, H-1), 4.59 (1H, d, J 11.3, CH2
(MPM)), 4.52 (1H, d, J 11.7, CH2
(MPM)), 4.09 (2H, s, H-2 and H-3), 3.97 (1H, d, J 7.6, H-5′a), 3.86 (1H, s, H-5a), 3.84 (1H, s, H-5b), 3.79 (3H, s, OCH3
(MPM)), 3.64 (1H, d, J 7.3, H-5′b), 3.37 (3H, s, OCH3), 2.14 (1H, dd, J 6.0 and 6.6, 5-OH); δC
(CDCl3) 159.5, 129.8, 129.4 and 113.9 (Ar), 105.2 (C-1), 85.6 (C-4), 78.3 and 77.4 (C-2 and C-3), 71.9 (CH2
(MPM)), 71.8 (C-5′), 58.8 (C-5), 55.5 and 55.3 (OCH3 and OCH3
(MPM)); MALDI-HRMS: m/z 319.1140 ([M + Na]+, C15H20O6Na+ calc. 319.1152). Similarly, reaction of furanoside 9b
(416 mg, 1.23 mmol) with sat. methanolic ammonia (20 cm3) followed by work-up and column chromatography (60%
(v/v) EtOAc in light petroleum) yielded furanoside 10b as a white solid material (328 mg, 90%). Rf 0.36 (EtOAc–light petroleum 75 ∶ 25, v/v); δH
(CDCl3) 7.28 (2H, d, J 7.3, Ar-H), 6.88 (2H, d, J 8.5, Ar-H), 4.97 (1H, s, H-1), 4.61 (1H, d, J 11.8, CH2
(MPM)), 4.54 (1H, d, J 11.3, CH2
(MPM)), 4.16 (1H, s, H-2), 3.98 (1H, d, J 8.0, H-5′a), 3.94 (1H, s, H-3), 3.86 (1H, d, J 7.8, H-5′b), 3.82–3.77 (5H, m, H-5 and OCH3
(MPM)), 3.47 (3H, s, OCH3), 2.11 (1H, t, J 6.0, 5-OH); δC
(CDCl3) 159.5, 129.5, 129.4 and 114.0 (Ar), 104.3 (C-1), 89.1 (C-4), 78.9 (C-3), 77.4 (C-2), 72.3 (C-5′), 71.8 (CH2
(MPM)), 58.7 (C-5), 56.3 (OCH3), 55.4 (OCH3
(MPM)); MALDI-HRMS: m/z 319.1149 ([M + Na]+, C15H20O6Na+ calc. 319.1152).
(1R,3SR,4S,7R)-3-Methoxy-7-(p-methoxybenzyloxy)-1-(p-methoxybenzyloxymethyl)-2,5-dioxabicyclo[2.2.1]heptane (11). Sodium hydride (240 mg, 60% suspension in mineral oil, 6.00 mmol) was added to anhydrous THF (3 cm3) and the resulting mixture was cooled to 0 °C. A solution of furanoside 10a
(667 mg, 2.25 mmol) in anhydrous THF (3 cm3) was added dropwise. The temperature of the mixture was allowed to increase to rt and stirring was continued for 15 min. p-Methoxybenzyl chloride (463 mg, 2.96 mmol) was added dropwise followed by tetrabutylammonium iodide (554 mg, 1.5 mmol), and the resulting mixture was stirred for 36 h at rt. The mixture was cooled to 0 °C and ice-cold H2O (10 cm3) was carefully added. Extraction was performed with EtOAc (2 × 20 cm3) and the combined organic phase was washed with brine (20 cm3), dried (Na2SO4), filtered and evaporated to dryness under reduced pressure. The residue was purified by column chromatography (35% EtOAc in light petroleum, containing 1% Et3N, v/v/v) to afford furanoside 11a as a colourless oil (806 mg, 86%). Rf 0.62 (EtOAc–light petroleum 50 ∶ 50, v/v); δH
(CDCl3) 7.26–7.20 (4H, m, Ar-H), 6.88–6.84 (4H, m, Ar-H), 4.79 (1H, s, H-1), 4.56 (1H, d, J 11.6, CH2
(MPM)), 4.53 (2H, s, CH2
(MPM)), 4.47 (1H, d, J 11.5, CH2
(MPM)), 4.07 and 4.04 (1H each, 2s, H-2 and H-3), 3.95 (1H, d, J 7.2, H-5′a), 3.79 (6H, s, 2 × OCH3
(MPM)), 3.75 (1H, d, J 7.8, H-5′b), 3.72 (1H, s, H-5a), 3.71 (1H, s, H-5b), 3.38 (3H, s, OCH3); δC
(CDCl3) 159.4, 159.3, 130.1, 129.8, 129.4, 129.3, 113.9 and 113.8 (Ar) 105.0 (C-1), 85.2 (C-4), 78.9 and 77.3 (C-2 and C-3), 73.4 (CH2
(MPM)), 72.4 (C-5′), 71.9 (CH2
(MPM)), 66.3 (C-5), 55.5 (OCH3), 55.3 (2 × OCH3
(MPM)); MALDI-HRMS: m/z 439.1726 ([M + Na]+, C23H28O7Na+ calc. 439.1727). Similarly, furanoside 10b
(278 mg, 0.94 mmol) was reacted with p-methoxybenzyl chloride (157 mg, 1.00 mmol) in the presence of NaH (102 mg, 60% suspension in mineral oil, 2.55 mmol), tetrabutylammonium iodide (185 mg, 0.5 mmol) and anhydrous THF (2 cm3) followed by work-up and column chromatography (50% EtOAc in light petroleum, containing 1% Et3N, v/v/v) to give furanoside 11b as a colourless oil (316 mg, 81%). Rf 0.31 (EtOAc–light petroleum 50 ∶ 50, v/v); δH
(CDCl3) 7.26–7.20 (4H, m, Ar-H), 6.87–6.85 (4H, m, Ar-H), 4.99 (1H, s, H-1), 4.60–4.46 (4H, m, 2 × CH2
(MPM)), 4.13 (1H, s, H-2), 3.95–3.87 (3H, m, H-3 and H-5′), 3.79 (6H, s, 2 × OCH3
(MPM)), 3.65 (2H, s, H-5), 3.48 (3H, s, OCH3); δC
(CDCl3) 159.5, 159.4, 129.9, 129.6, 129.5, 129.4, 113.9 and 113.8 (Ar), 104.1 (C-1), 88.6 (C-4), 79.3 (C-3), 77.3 (C-2), 73.4 (2 × CH2
(MPM)), 72.7 (C-5′), 71.7 (CH2
(MPM)), 65.4 (C-5), 56.4 (OCH3), 55.3 (OCH3
(MPM)); MALDI-HRMS: m/z 439.1713 ([M + Na]+, C23H28O7Na+ calc. 439.1727.
(2S,3R,4R)-4-Hydroxy-3-(p-methoxybenzyloxy)-4-(p-methoxybenzyloxymethyl)tetrahydrofuran-2-carbaldehyde (12). A solution of furanoside 11a
(7.19 g, 17.3 mmol) in 70% aqueous acetic acid (100 cm3
(v/v)) was stirred for 12 h at rt. The mixture was evaporated to dryness under reduced pressure and the residue was successively coevaporated with absolute ethanol (3 × 50 cm3) and toluene (3 × 50 cm3) and purified by column chromatography (40 (v/v) EtOAc in light petroleum) to give aldehyde 12 as a white solid material (5.90 g, 85%). Rf 0.33 (EtOAc–light petroleum 50 ∶ 50, v/v); δH
(CDCl3) 9.63 (1H, s, H-1), 7.18 (4H, d, J 8.5, Ar-H), 6.86 (2H, d, J 8.5, Ar-H), 6.85 (2H, d, J 8.7, Ar-H), 4.58 (1H, d, J 11.2, CH2
(MPM)), 4.49 (1H, d, J 11.4, CH2
(MPM)), 4.44 (1H, d, J 11.5, CH2
(MPM)), 4.43 (1H, d, J 11.4, CH2
(MPM)), 4.35 (1H, s, H-2), 3.94 (1H, d, J 9.2, H-5′a), 3.90 (1H, s, H-3), 3.88 (1H, d, J 9.1, H-5′b), 3.79–3.75 (7H, m, 2 × OCH3 and H-5a), 3.44 (1H, d, J 9.3, H-5b), 2.80 (1H, s, 4-OH); δC
(CDCl3) 203.6 (C-1), 159.5, 159.4, 129.6, 129.5, 129.3, 114.0 and 113.9 (Ar), 87.3 (C-2), 86.8 (C-3), 81.0 (C-4), 75.1 (C-5′), 73.4 (CH2
(MPM)), 71.6 (CH2
(MPM)), 67.6 (C-5), 55.3 (2 × OCH3
(MPM)); MALDI-HRMS: m/z 425.1570 ([M + Na]+, C22H26O7Na+ calc. 425.1570. Similarly, reaction of furanoside 11b
(3.19 g, 7.66 mmol) with 70% aqueous acetic acid (50 cm3) followed by evaporation, coevaporation and column chromatography afforded aldehyde 12
(2.47 g, 80%) with analytical data identical to those listed above.
(2R,3R,4R)-4-Hydroxy-2-[(S)-hydroxy(phenyl)methyl]-4-(p-methoxybenzyloxy)-3-(p-methoxybenzyloxymethyl)tetrahydrofuran (13a). Phenyl magnesium bromide (1.0 M solution in THF, 40 cm3, 40 mmol) was added dropwise during 15 min to a stirred solution of aldehyde 12
(2.90 g, 7.21 mmol) in anhydrous THF (25 cm3) at 0 °C. The temperature was allowed to rise to rt and stirring was continued for 12 h. The reaction mixture was evaporated to dryness under reduced pressure and the residue diluted with CH2Cl2
(50 cm3) and washed with sat. aq. NH4Cl (3 × 25 cm3). The organic phase was dried (Na2SO4), filtered, and concentrated to dryness under reduced pressure. The residue was purified by column chromatography (30%
(v/v) EtOAc in light petroleum) affording tetrahydrofuran 13a
(2.57 g, 74%) as a white solid material. Rf 0.33 (CH2Cl2–MeOH 95 ∶ 5, v/v); δH
(CDCl3) 7.40–7.29 (5H, m), 7.21 (2H, d, J 8.8), 6.90–6.84 (4H, m), 6.76 (2H, d, J 8.7), 4.73 (1H, dd, J 2.6 and 6.3, H-1′), 4.48 (2H, s, CH2
(MPM)), 4.09–4.05 (3H, m, H-2′ and CH2
(MPM)), 3.88 (1H, d, J 9.3, H-5″a), 3.81–3.75 (7H, m, 2 × OCH3 and H-5″b), 3.73–3.69 (2H, m, H-3′ and H-5′a), 3.49 (1H, d, J 9.4, H-5′b), 3.36 (1H, d, J 2.5, 1′-OH), 3.21 (1H, s, 4′-OH); δC
(CDCl3) 159.5, 159.4, 140.7, 129.7, 129.6, 129.5, 128.6, 128.1, 127.3, 114.0 and 113.8 (Ar), 89.4 (C-2′), 84.6 (C-3′), 81.9 (C-4′), 75.4 (C-5″), 74.8 (C-1′), 73.5 (CH2
(MPM)), 71.7 (CH2
(MPM)), 69.3 (C-5′), 55.4 (2 × OCH3); MALDI-HRMS: m/z 503.2016 ([M + Na]+, C28H32O7Na+ calc. 503.2040).
(2R,3R,4R)-4-Hydroxy-2-[(S)-hydroxy(1-pyrenyl)methyl]-4-(p-methoxybenzyloxy)-3-(p-methoxybenzyloxymethyl)tetrahydrofuran (13b). 1-Bromopyrene (6.27 g, 22.3 mmol) was added to a stirred mixture of magnesium turnings (930 mg, 38.3 mmol) and iodine (20 mg) in THF (20 cm3). The mixture was stirred at 40 °C for 1 h whereupon it was cooled to rt. A solution of aldehyde 12
(3.03 g, 7.53 mmol) in THF (10 cm3) was added slowly and the reaction mixture was stirred for 12 h. The crude product obtained after work-up (as described above for 13a) was purified by column chromatography (40%
(v/v) EtOAc in light petroleum) to give tetrahydrofuran 13b
(3.25 g, 71%) as a pale yellow solid. Rf 0.43 (EtOAc–light petroleum 50 ∶ 50, v/v); δH
(CDCl3) 8.33–8.27 (2H, m), 8.20–8.13 (3H, m), 8.05–7.99 (4H, m), 7.15 (2H, d, J 8.6), 6.82 (2H, d, J 8.7), 6.30 (2H, d, J 8.7), 6.20 (2H, d, J 8.3), 5.87 (1H, d, J 6.7, H-1′), 4.43 (2H, s, CH2
(MPM)), 4.41 (1H, m, H-2′), 4.01 (1H, d, J 9.3, H-5″a), 3.91 (1H, d, J 11.9, CH2
(MPM)), 3.86 (1H, d, J 9.4, H-5″b), 3.77 (3H, s, OCH3), 3.71–3.64 (4H, m, H-3′, H-5′a, OH, and CH2
(MPM)), 3.50 (1H, d, J 9.4, H-5′b), 3.44 (3H, s, OCH3), 3.32 (1H, br s, OH); δC
(CDCl3) 159.5, 158.9, 133.9, 131.4, 131.1, 130.7, 129.8, 129.7, 129.5, 129.2, 128.9, 128.5, 127.8, 127.7, 127.5, 126.0, 125.5, 125.3, 125.2, 125.1, 125.0, 124.9, 122.9, 113.9 and 113.3 (Ar), 89.5 (C-2′), 83.5 (C-3′), 82.0 (C-4′), 75.7 (C-5″), 73.4 (CH2
(MPM)), 71.3 and 71.0 (C-1′ and CH2
(MPM)), 69.3 (C-5′), 55.3 (OCH3), 55.0 (OCH3); MALDI-HRMS: m/z 627.2343 ([M + Na]+, C38H36O7Na+ calc. 627.2353).
(2S,3R,4R)-4-Hydroxy-2-[phenylcarbonyl]-4-(p-methoxybenzyloxy)-3-(p-methoxybenzyloxymethyl)tetrahydrofuran (14a). Dess Martin periodinane (2.01 g, 4.74 mmol) was added to the solution of compound 13a
(1.08 g, 2.25 mmol) in CH2Cl2
(10 cm3) and the reaction mixture was stirred for 12 h at rt. Diethyl ether (100 cm3) was added and the resulting mixture was poured into sat. aq. NaHCO3
(100 cm3, containing 2 g of Na2S2O3). The resulting mixture was slowly stirred until the white solid material dissolved. The phases were separated, and the aqueous layer was washed with ether (50 cm3). The combined organic phase was dried (Na2SO4), filtered and concentrated to dryness under reduced pressure. The residue was purified by column chromatography (25%
(v/v) EtOAc in light petroleum) to give compound 14a as a white solid material (823 mg, 77%). Rf 0.52 (EtOAc–light petroleum 50 ∶ 50, v/v); δH
(CDCl3) 7.91–7.88 (2H, m), 7.57–7.54 (1H, m), 7.41–7.36 (2H, m), 7.25–7.16 (4H, m) and 6.88–6.82 (4H, m, Ar-H), 5.14 (1H, d, J 2.3, H-2′), 4.59 (1H, d, J 11.2, CH2
(MPM)), 4.52–4.49 (3H, m, CH2
(MPM)), 4.25 (1H, d, J 2.3, H-3′), 3.97 (2H, s, H-5″), 3.79–3.77 (7H, m, H-5′a and 2 × OCH3), 3.57 (1H, d, J 9.4, H-5′b); δC
(CDCl3) 198.2 (C-1′), 159.5, 159.4, 135.5, 133.3, 129.8, 129.7, 129.6, 129.0, 128.5 and 114.0 (Ar), 86.7 (C-2′), 86.2 (C-3′), 81.9 (C-4′), 75.6 (C-5″), 73.5 and 72.7 (CH2
(MPM)), 69.0 (C-5′), 55.3 (2 × OCH3); MALDI-HRMS: m/z 501.1868 ([M + Na]+, C28H30O7Na+ calc. 501.1884).
(2S,3R,4R)-4-Hydroxy-2-[1-pyrenylcarbonyl]-4-(p-methoxybenzyloxy)-3-(p-methoxybenzyloxymethyl)tetrahydrofuran (14b). 3Å Molecular sieve powder (3.0 g) was activated by heating at 200 °C for 2 h under reduced pressure. After cooling to rt under an atmosphere of N2, pyridinium dichromate (1.5 g, 3.99 mmol) and anhydrous CH2Cl2
(20 cm3, freshly filtered through basic alumina) were added. Compound 13b
(1.51 g, 2.50 mmol) was added in one portion to the stirred solution and the sides of the flask were washed with CH2Cl2
(5 cm3). The resulting mixture was stirred for 2 h at rt and then filtered through 3Å molecular sieve powder on a glass filter. The plug was washed with CH2Cl2
(3 × 10 cm3) and the combined filtrate concentrated to dryness under reduced pressure. The resulting brownish residue was suspended in ethyl acetate (50 cm3), sonicated for 5 min, filtered through 3Å molecular sieve powder, washed with ethyl acetate (2 × 20 cm3) and concentrated to dryness under reduced pressure to give a residue which was used in the next step without further purification.
(2R,3R,4R)-4-Hydroxy-2-[(R)-hydroxy(phenyl)methyl]-4-(p-methoxybenzyloxy)-3-(p-methoxybenzyloxymethyl)tetrahydrofuran (15a). NaBH4
(330 mg, 8.71 mmol) was added at 0 °C to the solution of compound 14a
(1.93 g, 4.03 mmol) in a mixture of THF (10 cm3) and H2O (2.0 cm3). The resulting mixture was stirred at rt for 30 min whereupon ethyl acetate (50 cm3) and H2O (25 cm3) were added. The phases were separated and the organic phase was washed with sat. aqueous NaHCO3
(2 × 25 cm3), dried (Na2SO4), filtered and then concentrated to dryness under reduced pressure. The residue was purified by column chromatography (25%
(v/v) EtOAc in light petroleum) to give compound 15a
(432 mg, 22%) as a white solid material while elution with 30–35%
(v/v) EtOAc in light petroleum afforded stereoisomer 13a
(1.04 g, 54%). Data for 15a: Rf 0.73 (EtOAc–light petroleum 50 ∶ 50, v/v);δH
(CDCl3) 7.45–7.43 (2H, m), 7.39–7.35 (2H, m), 7.31–7.25 (1H, m), 7.22–7.19 (2H, m), 6.86–6.83 (2H, m), 6.75–6.72 (4H, m), 5.08 (1H, d, J 2.3, H-1′), 4.48 (2H, s, CH2
(MPM)), 4.17 (1H, dd, J 2.6 and 2.7, H-2′), 3.91 (1H, d, J 9.4, H-5′a), 3.83 (1H, s, OH), 3.78 (4H, s, OCH3 and H-3′), 3.76 (3H, s, OCH3), 3.75 (1H, d, J 9.2, H-5′b), 3.70 (1H, d, J 9.5, H-5″a), 3.68 (1H, d, J 10.9, CH2
(MPM)), 3.63 (1H, d, J 11.3, CH2
(MPM)), 3.58 (1H, s, OH), 3.51 (1H, d, J 9.7, H-5″b); δC
(CDCl3) 159.5, 159.2, 140.7, 129.8, 129.6, 129.2, 128.6, 127.6, 126.1, 114.0 and 113.7 (Ar), 90.9 (C-2′), 82.1 (C-3′), 81.2 (C-4′), 75.3 (CH2
(MPM)), 73.6 (CH2
(MPM)), 73.1 (C-1′), 71.1 (C-5″), 69.0 (C-5′), 55.3 (OCH3
(MPM)); MALDI-HRMS: m/z 503.2018 ([M + Na]+, C28H32O7Na+ calc. 503.2040).
(2R,3R,4R)-4-Hydroxy-2-[(R)-hydroxy(1-pyrenyl)methyl]-4-(p-methoxybenzyloxy)-3-(p-methoxybenzyloxymethyl)tetrahydrofuran (15b). The crude 14b
(∼1.4 g) was reacted with NaBH4
(166 mg, 4.49 mmol) in a 1 ∶ 4 mixture of H2O–THF (10 cm3). Work-up (as described above for 15a) followed by column chromatography (30–35%
(v/v) EtOAc in light petroleum) furnished compound 15b
(246 mg, 16% from 13b, two steps) as a white solid material while elution with 35–40%
(v/v) EtOAc in light petroleum furnished the stereoisomer 13b
(507 mg, 34%) as a white solid material. Data for 15b: Rf 0.53 (EtOAc–light petroleum 50 ∶ 50, v/v); δH
(CDCl3) 8.27 (1H, J 8.3), 8.08–8.02 (4H, m), 7.93–7.86 (4H, m), 7.16–7.10 (2H, m), 6.76–6.73 (2H, m), 6.14–6.11 (2H, m), 5.95 (3H, d, J 8.4, Ar-H and H-1′), 4.42–4.39 (3H, m, H-2′ and CH2
(MPM)), 4.29 (1H, br s, OH), 3.93 (1H, d, J 9.3, H-5″a), 3.83–3.60 (7H, m, H-3′, H-5″b, OH, OCH3 and CH2
(MPM)), 3.45–3.42 (4H, m, OCH3 and CH2
(MPM)), 3.22 (1H, d, J 11.7, H-5′a), 3.09 (1H, d, J 11.6, H-5′b); δC
(CDCl3) 159.4, 158.7, 133.6, 131.3, 130.8, 130.6, 129.7, 129.6, 129.1, 128.5, 127.9, 127.5, 127.3, 127.2, 126.0, 125.4, 125.2, 124.8, 124.6, 123.6, 121.9, 113.9 and 113.1 (Ar), 89.4 (C-2′), 82.7 (C-3′), 81.3 (C-4′), 75.4 (C-5″), 73.5 (CH2
(MPM)), 70.8 and 70.6 (C-1′ and CH2
(MPM)), 69.0 (C-5′), 55.3 (OCH3), 55.0 (OCH3); MALDI-HRMS: m/z 627.2329 ([M + Na]+, C38H36O7Na+ calc. 627.2353).
(1R,3S,4S,7R)-7-(p-Methoxybenzyloxy)-1-(p-methoxybenzyloxymethyl)-3-phenyl-2,5-dioxabicyclo[2.2.1]heptane (16a). N,N,N′,N′-Tetramethylazodicarboxamide (TMAD, 277 mg, 1.61 mmol) was added in one portion to a stirred solution of compound 15a
(492 mg, 1.02 mmol) and tributylphosphine (PBu3, 328 mg, 1.62 mmol) in anhydrous benzene (10 cm3) at 0 °C. The mixture was stirred 12 h at rt and was then diluted with diethyl ether (50 cm3). Washing was performed successively with sat. aq. NH4Cl (2 × 20 cm3) and brine (25 cm3) and the separated organic phase was dried (Na2SO4), filtered and concentrated to dryness under reduced pressure. The residue was purified by column chromatography (15–20%
(v/v) EtOAc in light petroleum) to give compound 16a as a white solid material (461 mg, 97%). Rf 0.75 (EtOAc–light petroleum 50 ∶ 50, v/v); δH
(CDCl3) 7.36–7.34 (4H, m), 7.30–7.24 (5H, m), 6.89–6.86 (4H, m), 5.15 (1H, s, H-1′), 4.63 (1H, d, J 11.8, CH2
(MPM)), 4.62 (1H, d, J 12.1, CH2
(MPM)), 4.56 (2H, d, J 11.4, CH2
(MPM)), 4.34 and 4.28 (1H each, 2s, H-2′ and H-3′), 4.00 (1H, d, J 7.6, H-5″a), 3.94 (1H, d, J 7.8, H-5″b), 3.80 (3H, s, OCH3), 3.79 (3H, s, OCH3), 3.78 (2H, s, H-5′); δC
(CDCl3) 159.5, 159.4, 138.7, 130.1, 129.9, 129.5, 129.4, 128.2, 127.5, 126.1, 113.9 and 113.8 (Ar), 87.4 (C-4′), 82.2 (C-1′), 80.8 and 79.4 (C-2′ and C-3′), 73.9 (C-5″), 73.5 (CH2
(MPM)), 71.7 (CH2
(MPM)), 65.9 (C-5′), 55.4 (2 × OCH3); MALDI-HRMS: m/z 485.1921 ([M + Na]+, C28H30O6Na+ calc. 485.1935).
(1R,3S,4S,7R)-7-(p-Methoxybenzyloxy)-1-(p-methoxybenzyloxymethyl)-3-(1-pyrenyl)-2,5-dioxabicyclo[2.2.1]heptane (16b). Reaction of compound 15b
(401 mg, 0.66 mmol) in the presence of TMAD (170 mg, 0.98 mmol), PBu3
(200 mg, 0.99 mmol) and benzene (10 cm3) followed by work-up (as described above for 16a) and column chromatography (20%
(v/v) EtOAc in light petroleum) afforded compound 16b as a pale yellow solid (342 mg, 88%). Rf 0.53 (EtOAc–light petroleum 50 ∶ 50, v/v); δH
(CDCl3) 8.42 (1H, d, J 8.0), 8.21 (1H, d, J 7.9), 8.15 (2H, d, J 7.9), 8.07 (1H, d, J 9.2), 8.04–7.95 (4H, m), 7.33 (2H, d, J 8.5), 7.29 (2H, d, J 8.4), 6.90 (2H, d, J 8.7), 6.89 (2H, d, J 8.4), 6.17 (1H, s, H-1′), 4.72 (1H, s, H-2′), 4.71–4.61 (4H, m, 2 × CH2
(MPM)), 4.52 (1H, s, H-3′), 4.21 (1H, d, J 7.6, H-5″a), 4.07 (1H, d, J 7.8, H-5″b), 3.93 (1H, d, J 11.4, H-5′a), 3.88 (1H, d, J 11.4, H-5′b), 3.80 (6H, s, 2 × OCH3); δC
(CDCl3) 159.5, 159.4, 131.9, 131.4, 130.9, 130.6, 130.2, 129.9, 129.6, 129.5, 129.4, 129.3, 127.8, 127.7, 127.6, 127.4, 127.1, 125.9, 125.4, 125.1, 124.9, 124.8, 124.2, 121.9, 114.0 and 113.9 (Ar), 87.5 (C-4′), 81.1 (C-3′), 80.6 (C-1′), 79.3 (C-2′), 73.8 (C-5″), 73.6 (CH2
(MPM)), 71.9 (CH2
(MPM)), 66.1 (C-5′), 55.4 (2 × OCH3); MALDI-HRMS: m/z 609.2240 ([M + Na]+, C38H34O6Na+ calc. 609.2247).
(1R,3S,4S,7R)-7-Hydroxy-1-hydroxymethyl-3-phenyl-2,5-dioxabicyclo[2.2.1]heptane (17a). 2,3-Dichloro-5,6-dicyanoquinone (DDQ, 417 mg, 1.84 mmol) was added at rt to a stirred solution of compound 16a
(461 mg, 1.0 mmol) in CH2Cl2
(7 cm3) containing a small amount of H2O (0.3 cm3). Immediate appearance of a deep greenish-black colour which slowly faded into pale brownish-yellow was observed. The reaction mixture was vigorously stirred for 4 h. The precipitate was removed by filtration through a short pad of silica gel which was washed with EtOAc (2 × 25 cm3). The combined filtrate was washed successively with sat. aq. NaHCO3
(25 cm3) and brine (25 cm3). The separated organic phase was dried (Na2SO4), filtered and concentrated to dryness under reduced pressure. The residue was purified by column chromatography (65%
(v/v) EtOAc in light petroleum) to give compound 17a as a white solid material (140 mg, 63%). Rf 0.38 (EtOAc–light petroleum 75 ∶ 25, v/v); δH
(CDCl3–CD3OD; 5 ∶ 1) 7.30–7.21 (5H, m, Ar-H), 5.12 (1H, s, H-1′), 4.38 (1H, s, H-2′), 4.20 (1H, s, H-3′), 3.91 (1H, d, J 8.1, H-5″a), 3.83–3.80 (3H, m, H-5′ and H-5″b), 2.99 (1H, s, OH); δC
(CDCl3–CD3OD, 5 ∶ 1) 138.4, 128.3, 127.5, 125.8, 88.3, 81.9, 81.8, 74.4, 72.7, 58.2; MALDI-HRMS: m/z 245.0791 ([M + Na]+, C12H14O4Na+ calc. 245.0784).
(1R,3S,4S,7R)-7-Hydroxy-1-hydroxymethyl-3-(1-pyrenyl)-2,5-dioxabicyclo[2.2.1]heptane (17b). Reaction of compound 16b
(326 mg, 0.57 mmol) with DDQ (452 mg, 1.99 mmol) in a mixture of CH2Cl2
(8 cm3) and H2O (0.5 cm3) at rt followed by work-up (as described above for 17a) and column chromatography (2%
(v/v) MeOH in CH2Cl2) furnished compound 17b as a white solid material (100 mg, 52%). Rf 0.34 (EtOAc–light petroleum 75 ∶ 25 v/v); δH
((CD3)2SO), 8.43 (1H, d, J 8.1), 8.34–8.29 (4H, m), 8.22 (1H, d, J 9.5), 8.17–8.08 (3H, m), 6.22 (1H, s, H-1′), 5.68 (1H, d, J 4.4, 3′-OH), 4.96 (1H, dd, J 5.5 and 6.1, 5′-OH), 4.54 (1H, d, J 4.8, H-3′), 4.52 (1H, s, H-2′), 4.03 (1H, d, J 7.8, H-5″a), 3.91 (1H, d, J 7.7, H-5″b), 3.88 and 3.86 (1H each, 2s, H-5′); δC
((CD3)2SO) 133.7, 130.8, 130.1, 129.9, 127.4, 127.3, 126.8, 126.1, 125.2, 125.1, 124.4, 124.2, 124.0, 123.7 and 122.6 (Ar), 88.5 (C-4′), 81.7 (C-2′), 79.2 (C-1′), 74.2 (C-3′), 72.4 (C-5″), 58.2 (C-5′); MALDI-HRMS: m/z 369.1106 ([M + Na]+, C22H18O4Na+ calc. 369.1097).
(1S,3S,4S,7R)-1-(4,4′-Dimethoxytrityloxymethyl)-7-hydroxy-3-phenyl-2,5-dioxabicyclo[2.2.1]heptane (18a). 4,4′-Dimethoxytrityl chloride (195 mg, 0.58 mmol) was added in one portion to a stirred solution of the compound 17a
(101 mg, 0.45 mmol) in anhydrous pyridine (1.5 cm3). After stirring for 12 h at rt, methanol (0.1 cm3) was added and the resulting mixture was concentrated to dryness under reduced pressure. The residue was dissolved in CH2Cl2
(20 cm3) and washed with sat. aq. NaHCO3
(2 × 10 cm3). The separated organic phase was dried (Na2SO4), filtered, concentrated to dryness under reduced pressure and coevaporated with toluene (2 × 2 cm3). The residue was purified by column chromatography (50% ethyl acetate in light petroleum, containing 0.5% Et3N (v/v/v)) to afford compound 18a as a white solid material (210 mg, 88%). Rf 0.59 (EtOAc–light petroleum 75 ∶ 25 v/v); δH
(CDCl3) 7.39 (2H, d, J 7.5), 7.38–7.15 (12H, m), 6.77 (4H, d, J 8.9), 5.15 (1H, s, H-1′), 4.34 (1H, d, J 5.1, H-3′), 4.21 (1H, s, H-2′), 3.94 (2H, s, H-5″), 3.71 (6H, s, 2 × OCH3), 3.48 (1H, d, J 10.6, H-5′a), 3.42 (1H, d, J 10.4, H-5′b), 2.30 (1H, d, J 5.6, 3′-OH); δC
(CDCl3) 158.7, 144.6, 138.7, 135.8, 135.6, 130.2, 128.4, 128.2, 128.1, 127.5, 127.0, 125.9 and 113.4 (Ar), 86.7 and 86.6 (C-4′ and –CAr3), 81.9 and 81.8 (C-1′ and C-2′), 76.4 (C-3′), 73.2 (C-5″), 61.3 (C-5′), 55.3 (2 × OCH3), MALDI-HRMS: m/z 547.2095 ([M + Na]+, C33H32O6Na+ calc. 547.2091).
(1S,3S,4S,7R)-1-(4,4′-Dimethoxytrityloxymethyl)-7-hydroxy-3-(1-pyrenyl)-2,5-dioxabicyclo[2.2.1]heptane (18b). Reaction of compound 17b
(57 mg, 0.16 mmol) with DMTCl (64 mg, 0.19 mmol) in anhydrous pyridine (1 cm3) followed by work-up (as described above for 18a) and column chromatography (35% EtOAc in light petroleum, containing 0.5% Et3N (v/v/v)) afforded compound 18b as a yellowish solid material (58 mg, 54%). Rf 0.72 (EtOAc–light petroleum 75:25, v/v); δH
(CDCl3) 8.38 (1H, d, J 8.0), 8.16 (1H, d, J 8.0), 8.08 (2H, d, J 8.1), 8.00–7.90 (5H, m), 7.45 (2H, d, J 8.5), 7.34 (4H, d, J 7.1), 7.32–7.16 (3H, m), 6.79 (4H, d, J 8.9), 6.11 (1H, s, H-1′), 4.64 (1H, s, H-2′), 4.57 (1H, br s, H-3′), 4.20 (1H, d, J 8.4, H-5″a), 4.01 (1H, d, J 8.4, H-5″b), 3.71 (6H, s, 2 × OCH3), 3.60 (1H, d, J 10.4, H-5′a), 3.54 (1H, d, J 10.0, H-5′b), 2.42 (1H, br s, 3′-OH); δC
(CDCl3) 158.7, 144.7, 135.8, 135.7, 131.7, 131.4, 131.0, 130.6, 130.2, 128.2, 128.1, 127.7, 127.6, 127.5, 127.2, 127.0, 126.0, 125.7, 125.4, 125.2, 125.0, 124.9, 124.8, 123.9, 122.0 and 113.4 (Ar), 86.9 (C-4′), 86.6 (CAr3), 81.8 (C-2′), 80.3 (C-1′), 76.6 (C-3′), 73.0 (C-5″), 61.4 (C-5′), 55.3 (2 × OCH3); ESI-MS: m/z 671.5 [M + Na]˙
(1S,3S,4S,7R)-7-[2-Cyanoethoxy(diisopropylamino)phosphinoxy]-1-(4,4′-dimethoxytrityloxymethyl)-3-phenyl-2,5-dioxabicyclo[2.2.1]heptane (19a). 2-Cyanoethyl N,N-diisopropylphosphoramidochloridite (50 mg, 0.21 mmol) was added dropwise to a stirred solution of the compound 18a
(100 mg, 0.19 mmol) and N,N-diisopropylethylamine (0.2 cm3) in anhydrous CH2Cl2
(1.0 cm3) at rt. After stirring for 6 h at rt, the reaction mixture was diluted with EtOAc (20 cm3, containing 0.5% Et3N, v/v). Washing was performed with sat. aq. NaHCO3
(2 × 10 cm3) and the separated organic phase was dried (Na2SO4), filtered and concentrated to dryness under reduced pressure. The residue was purified by column chromatography (20% EtOAc in n-hexane containing 0.5% Et3N (v/v/v)) to give phosphoramidite 19a as a white solid material (78 mg, 57%). Rf 0.61 (EtOAc–light petroleum 75 ∶ 25, v/v); δP
(CDCl3) 149.2.
(1S,3S,4S,7R)-7-[2-Cyanoethoxy(diisopropylamino)phosphinoxy]-1-(4,4′-dimethoxytrityloxymethyl)-3-(1-pyrenyl)-2,5-dioxabicyclo[ 2.2.1]heptane (19b). Reaction of compound 18b
(65 mg, 0.1 mmol) with 2-cyanoethyl N,N-diisopropylphosphoramidochloridite (29 mg, 0.12 mmol) in the presence of N,N-diisopropylethylamine (0.1 cm3) and anhydrous CH2Cl2
(1.0 cm3) followed by work-up (as described above for the compound 19a) and column chromatography (25% EtOAc in n-hexane containing 0.5% Et3N (v/v/v)) afforded phosphoramidite 19b as a white solid material (36 mg, 42 %). Rf 0.75 (EtOAc–light petroleum 50:50 v/v); δP
(CDCl3) 149.3, 149.2.
Synthesis, deprotection and purification of oligonucleotides
All oligonucleotides were prepared using the phosphoramidite approach on a Biosearch 8750 DNA synthesizer in 0.2 µmol scale on CPG solid supports (BioGenex). The stepwise coupling efficiencies for phosphoramidites 19a and 19b
(10 min coupling time) and for unmodified deoxynucleoside phosphoramidites (with standard 2 min coupling time) were >99% using 1H-tetrazole as activator (pyridine hydrochloride was used as activator when coupling 19b). After standard deprotection and cleavage from the solid support using 32% aqueous ammonia (12 h, 55 °C), the oligonucleotides were purified by precipitation from ethanol. The composition of the oligomers was verified by MALDI-MS analysis and the purity (>80%) by capillary gel electrophoresis. MALDI-MS m/z
([M − H]−, found/calc.): ON3, 2732/2733; ON4, 2854/2857; ON5, 2789/2789; ON6, 2909/2913.Thermal denaturation studies
The thermal denaturation experiments were performed on a Perkin-Elmer UV/VIS spectrometer fitted with a PTP-6 Peltier temperature-programming element using a medium salt buffer (10 mM sodium phosphate, 100 mM sodium chloride, 0.1 mM EDTA, pH 7.0) or a high salt buffer (10 mM sodium phosphate, 700 mM sodium chloride, 0.1 mM EDTA, pH 7.0) and concentrations of 1.0 µM of the two complementary strands (assuming identical extinction coefficients of the thyminyl, phenyl and pyrenyl moieties at 260 nm). During melting experiemnts, the absorbance was monitored at 260 nm while the temperature was raised at a rate of 1 °C min−1. The melting temperatures (Tm values) of the duplexes were determined as the maximum of the first derivatives of the melting curves obtained.Fluorescence measurements
Fluorescence emission intensities were recorded in buffer (10 mM sodium phosphate, 700 mM sodium chloride, 0.1 mM EDTA, pH 7.0) using 1.5 × 10−7 M concentrations of each strand (assuming identical extinction coefficients of the thyminyl, phenyl and pyrenyl moieties at 260 nm). All measurements were carried out with argon-saturated solutions at 19 ± 1 °C. Duplexes were heated to 70 °C and cooled to 0 °C before degassing and fluorescence measurements. Excitation wavelength (λex) was 280 nm. Emission slit width and excitation slit width were 4.0. Fluorescence intensities of band I (II) and band III (IIII) were measured at 383 nm and 401 nm, respectively.Acknowledgements
We thank the Danish National Research Foundation for financial support. Ms Britta M. Dahl is thanked for oligonucleotide synthesis and Dr Michael Meldgaard, Exiqon A/S, for MALDI-MS analyses.References
- D. Loakes, Nucleic Acids Res., 2001, 29, 2437 CrossRef CAS.
- R. Nichols, P. C. Andrews, P. Zhang and D. E. Bergstrom, Nature, 1994, 369, 492 CrossRef CAS.
- D. Loakes and D. M. Brown, Nucleic Acids Res., 1994, 22, 4039 CAS.
- M. Berger, Y. Wu, A. K. Ogawa, D. L. McMinn, P. G. Schultz and F. E. Romesberg, Nucleic Acids Res., 2000, 28, 2911 CrossRef CAS.
- F. Seela and H. Debelak, Nucleic Acids Res., 2000, 28, 3224 CrossRef CAS.
- T. J. Matray and E. T. Kool, J. Am. Chem. Soc., 1998, 120, 6191 CrossRef CAS.
- B. R. Babu and J. Wengel, Chem. Commun., 2001, 2114 RSC.
- B. R. Babu, A. K. Prasad, S. Trikha, N. Thorup, V. S. Parmar and J. Wengel, J. Chem. Soc., Perkin Trans. 1, 2002, 2509 RSC.
- A. A. Koshkin, S. K. Singh, P. Nielsen, V. K. Rajwanshi, R. Kumar, M. Meldgaard, C. E. Olsen and J. Wengel, Tetrahedron, 1998, 54, 3607 CrossRef CAS.
- M. D. Sørensen, L. Kværnø, T. Bryld, A. E. Håkansson, B. Verbeure, G. Gaubert, P. Herdewijn and J. Wengel, J. Am. Chem. Soc., 2002, 124, 2164 CrossRef CAS.
- K. M. E. Nielsen, M. Petersen, A. E. Håkansson, J. Wengel and J. P. Jacobsen, Chem. Eur. J., 2002, 8, 3001 CrossRef CAS.
- S. Obika, Y. Hari, K. Morio and T. Imanishi, Tetrahedron Lett., 2000, 41, 215 CrossRef CAS.
- S. Obika, Y. Hari, K. Morio and T. Imanishi, Tetrahedron Lett., 2000, 41, 221 CrossRef CAS.
- Y. Hari, S. Obika, M. Sakaki, K. Morio, Y. Yamagata and T. Imanishi, Tetrahedron, 2002, 58, 3051 CrossRef CAS.
- V. K. Rajwanshi, A. E. Håkansson, M. D. Sørensen, S. Pitsch, S. K. Singh, R. Kumar, P. Nielsen and J. Wengel, Angew. Chem., Int. Ed., 2000, 39, 1656 CrossRef CAS.
- K. Yamana, R. Iwase, S. Furutani, H. Tsuchida, H. Zako, T. Yamaoka and A. Murakami, Nucleic Acids Res., 1999, 27, 2387 CrossRef CAS.
- V. A. Korshun, D. A. Stetsenko and M. J. Gait, J. Chem. Soc., Perkin Trans. 1, 2002, 1092 RSC.
- U. B. Christensen and E. B. Pedersen, Nucleic Acids Res., 2002, 30, 4918 CrossRef CAS.
- A. Mahara, R. Iwase, T. Sakamoto, T. Yamaoka, K. Yamana and A. Murakami, Bioorg. Med. Chem., 2003, 11, 2783 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of the 13C NMR spectra of compounds 1–4, 5–11
(of both isomers), 12,13–18
(of phenyl and pyrenyl derivatives) and copies of the 31P NMR spectra of compounds 19a and 19b. See http://www.rsc.org/suppdata/ob/b3/b310719a/ |
| ‡ A research centre funded by the Danish National Research Foundation for studies on nucleic acid chemical biology. |
| § We have defined LNA as an oligonucleotide containing one or more 2′-O,4′-C-methylene-β-D-ribofuranosyl nucleotide monomer(s)
(ref. 9). Analogously, α-L-LNA has been defined as an oligonucleotide containing one or more 2′-O,4′-C-methylene-α-L-ribofuranosyl nucleotide monomer(s)
(ref. 10). |
| ¶ Despite the apparent differences in the furanose conformations between an LNA monomer and an α-L-LNA monomer, also the furanose conformation of an α-L-LNA monomer is of the N-type (C3′-endo, 3E) because of its L-configuration. For further information about the conformations of nucleotides, see Eur. J. Biochem. 1983, 131, 9 (“Abbreviations and Symbols for the Description of Conformations of Polynucleotide Chains”; IUPAC-IUB Joint Commission on Biochemical Nomenclature). |
|
| This journal is © The Royal Society of Chemistry 2004 |
Click here to see how this site uses Cookies. View our privacy policy here.