Stabilisation of the transition state of phosphodiester bond cleavage within linear single-stranded oligoribonucleotides
Ulla
Kaukinen
*ab,
Harri
Lönnberg
a and
Mikael
Peräkylä
b
aDepartment of Chemistry, University of Turku, FIN-20014, Turku, Finland
bDepartment of Chemistry, University of Kuopio, FIN-70211, Kuopio, Finland
First published on 24th November 2003
Abstract
The effect of base sequence on the stability of the transition state (TS) of phosphodiester bond cleavage within linear single-stranded oligoribonucleotides has been studied in order to better understand why the reactivity of some phosphodiester bonds is enhanced compared to an unconstrained linkage. Molecular dynamics simulations of 3.0 ns were carried out for 14 oligonucleotides that contain in the place of the scissile phosphodiester bond a phosphorane structure mimicking the TS of the bond cleavage. The hydrolytic stability of the same oligonucleotides had previously been reported. Both the non-bridging oxyanions and the leaving 5′-oxygen of the pentacoordinated phosphorane moiety were observed to form hydrogen bonds with solvent water molecules in a similar way with all the compounds studied. In addition, water mediated hydrogen bonds between the phosphorane non-bridging oxyanions and the bases of the 3′-flanking sequence were detected with some of the compounds, but not with the most labile ones. Hence, it seems that the enhanced cleavage of some internucleosidic linkages does not result from the TS stabilisation by hydrogen bonding. With heterooligomers, the stacking of bases next to the cleavage site was observed to be enhanced on going from the initial state to the TS, whereas within uracil homooligomer, having initially negligible stacking, no change in the magnitude of stacking was seen. Accordingly, while strong stacking in the initial state is known to retard the phosphodiester bond cleavage, it may in the TS accelerate the reaction. Therefore, enhanced stacking on going from the initial state to transition state appears to be a factor that markedly contributes to the hydrolytic stability of phosphodiester bonds within oligonucleotides and may, at least partly, explain accelerated cleavage compared to fully unconstrained bonds, such as those in polyuridylic acid.
Introduction
The cleavage of RNA phosphodiester bonds by intramolecular transesterification (Scheme 1) has attracted considerable interest in the scientific community since the 1980s, when it was discovered that, in addition to protein enzymes, RNA is capable of catalysis.1,2 The desire to develop artificial nucleases,3 which are hoped to offer new methods to treat, for example, cancer, viral infections and hereditary diseases, has also been a motivation to learn and understand the factors that govern the reactivity.4–8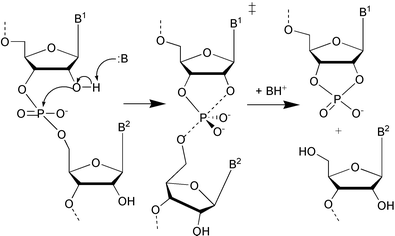 | ||
| Scheme 1 | ||
Under physiological conditions (pH ∼ 7) hydroxide ion catalysed cleavage of phosphodiester bonds is the predominant reaction mechanism (Scheme 1).4 The 2′-oxyanion, which is deprotonated in a rapid pre-equilibrium step, attacks the adjacent phosphate group, resulting in formation of a dianionic pentacoordinated species. The subsequent cleavage of the exocyclic P–O-bond leads to strand scission. According to the rules proposed by Westheimer,9 the rupture of the P–5′O bond is allowed only when the attacking and departing groups are co-linear with the phosphorus atom. It is not quite clear, whether the reaction is a two step process with a marginally stable dianionic phosphorane intermediate or whether the reaction is more like a concerted process going via a phosphorane-like transition state (TS). According to ab initio quantum mechanical (QM) calculations, cyclic dianionic phosphoranes show only a very shallow energy minimum in the gas phase, if any,10–15 but solvation has been proposed to stabilise the structure.16,17 The βlg value of −0.54 obtained with uridine 3′-aryl phosphates18 suggests that both the P–O2′ bond formation and P–O5′ bond cleavage are only moderately advanced in the TS, giving support to a concerted mechanism for the cleavage of aryl esters. The highly negative βlg value (−1.28) of the hydroxide ion catalysed cleavage of uridine 3′-alkylphosphates,19 however, proposes that with alkyl esters the departure of leaving alkoxide ion is almost complete in the TS. Accordingly, the reaction closely resembles a two step process, although the intermediate still may be extremely short-lived. Regardless of whether the reaction proceeds by a concerted one-step mechanism or via a marginally stable phosphorane intermediate, the overall rate-limiting step in the cleavage of the internucleosidic phosphodiester linkage involves the rupture of the exocyclic P–O bond.4,8
The hydrolytic stability of phosphodiester bonds has been observed to be strongly influenced by the molecular environment.5,20,21 Interestingly, not only defined secondary structures but also the base sequence within linear single-stranded RNA molecules has been found to exert a considerable influence on the phosphodiester bond reactivity.20,22–25 The base sequence has been observed to both accelerate and retard the cleavage of internucleosidic linkages by more than one order of magnitude compared to the rupture of an unconstrained linkage.20 In addition to the neighbouring bases of the scissile phosphodiester bond, those further apart have been found to contribute to the cleavage rate in a complicated way.20,24,25
The structure of a single-stranded RNA molecule is mainly determined by stacking interactions between bases. The stacking propensity of bases is known to be complicated, so that the identity of both the flanking bases and those further apart have been found to influence the stacking of two adjacent base moieties in a cooperative manner.26 Therefore, it is obvious that the base stacking has some contribution to the reactivity differences observed within linear single-stranded oligonucleotides. Comparison of the stacking tendency of bases within linear single-stranded heterooligomers26 with the experimental cleavage rates20 shows that the stacking of bases across the cleavage site clearly plays a role. It has been found that the rate retardations compared to an isolated phosphodiester bond may, to a significant extent, be attributed to the strong stacking of bases across the cleavage site.26 Since the co-linear orientation of attacking and leaving groups at the phosphorus, which according to Westheimer's rules9 is a prerequisite for the cleavage reaction, is not initially possible for conformational reasons, it is clear that the increasing rigidity the structure in the vicinity of the scissile linkage retards the reorientation required. This is then observed as a slower reaction. In contrast to the rate retardations, the origin of rate accelerations compared to unconstrained linkages has still remained as an open question. It has been observed that in the initial state the base–base interactions over the very labile linkages are weaker than those of hydrolytically very stable ones, but, however, clearly stronger than within the reference compound polyU exhibiting negligible stacking.26 Since it is unlikely that rate accelerations within single-stranded compounds could result from a favourable in-line geometry of reacting groups in the initial state,20 as suggested to be case with defined secondary structures,27 the TS within linear single-stranded compounds must be somehow stabilised. This stabilisation has frequently been speculated to originate from hydrogen bonding.20,24,28 It has been discussed that a hydrogen bond network, either direct or water mediated, between the 5′-linked base (especially the adenine base) and the phosphorane non-bridging oxyligands, or the attacking or leaving oxygen atoms, facilitates the proton transfer to the leaving group, and hence accelerates the cleavage.20,24,28 In particular, the protonation of the developing 5′-oxyanion, which lowers the overall energy barrier of the TS, has been thought to be important.4,6,20 In addition to the TS stabilisation by hydrogen bonding, it has recently been proposed26 that enhanced base stacking within the flanking sequences of the cleavage site might also stabilise the TS structure within oligomers. The underlying idea is that since weak stacking in one part of a molecule seems to be compensated by strong stacking elsewhere in the molecule,26 the complete loss of base stacking at the cleavage site could induce enhanced stacking of the flanking bases at the cleavage site, and accordingly, stabilise the TS. Within fully flexible reference compounds, this kind of TS stabilisation most obviously is not possible because of inherent negligible stacking tendency. In other words, when strong stacking in the initial state retards the reaction, enhanced stacking in the TS might, in turn, accelerate the reaction.
The knowledge about the overall structure of the RNA chain in the vicinity of the cleavage site is, scarce, and no quantitive data about TS stabilisations are available. Theoretical investigations of transition states have so far had their focus mainly on ab inito QM and density functional theory calculations of small model compounds of pentacoordinated phosphorane.8,29–32 In the present work, the TS structures of phosphodiester bond cleavage within linear single-stranded RNA molecules have been studied by the means of molecular dynamics (MD) simulations. The aim is to give an insight into both the TS structures of linear single-stranded oligonucleotides as well as into interactions which stabilise TS. This information is essential in order to better understand structural factors that contribute to the reactivity of internucleosidic linkages. MD simulations were performed for 14 linear single-stranded oligonucleotides, that contained in place of the scissile phosphodiester bond a phosphorane structure mimicking the TS of the bond cleavage. The inherent reactivity of the corresponding oligonucleotides has recently been reported.20 The selected compounds experience both rate-accelerations and retardations compared to a fully flexible reference compound, which is one of the compounds studied here. To our knowledge, no such data of the TS structures of linear single-stranded oligonucleotides are available.
Results
The oligonucleotides studied
The compounds studied (1–14), 12 and 13-mer chimeric ribo/2′-O-methylribo oligonucleotides, are listed in Table 1. All oligonucleotides contain only one ribo unit, the rest of the nucleosides being 2′-O-methylated. Accordingly, in such compounds only one of the phosphodiester bonds is able to undergo an intramolecular transesterification reaction, which has allowed an accurate determination of the cleavage rate of one particular bond in different molecular environments.20 The cleavage rates of these compounds, in the absence of any cofactors, has recently been reported (Table 1).20 Chimeric ribo/2′-O-methylribo oligonucleotides were chosen as model compounds because they are known to resemble natural RNA reasonably well.33,34 Evidence that compounds 1–14 really exist in a linear single-stranded form has been presented earlier.20| Compound | Sequence | k/10−7 s−1a |
|---|---|---|
| Bold letters refer to ribonucleosides, the other nucleosides being 2′-O-methylated. The position of the strand scission is indicated with a vertical line. a In 0.1 M CHES buffer, pH 8.5(I = 0.1 M with NaNO3).b No reaction in three months. | ||
| 1 | 5′GGGUAU|AAGUGC3′ | 14.6 ± 0.2 |
| 2 | 5′GGGUAA|AAGUGC3′ | <0.2b |
| 3 | 5′GGGUAC|AAGUGC3′ | 0.3 ± 0.1 |
| 4 | 5′GGGUAG|AAGUGC3′ | 0.5 ± 0.1 |
| 5 | 5′GGGUUU|AAGUGC3′ | 31.7 ± 0.6 |
| 6 | 5′GGGUAU|AUGUGC3′ | 43.8 ± 0.2 |
| 7 | 5′GGGUAU|AAGUUC3′ | 30.1 ± 0.1 |
| 8 | 5′GUGUAU|AAGUGC3′ | 1.2 ± 0.1 |
| 9 | 5′GUGUAC|AAGUGC3′ | 1.4 ± 0.1 |
| 10 | 5′GGGU|AUAAGUGC3′ | 0.9 ± 0.1 |
| 11 | 5′GGGUAUA|AGUGC3′ | <0.2b |
| 12 | 5′CCCCAAU|AACCCC3′ | <0.2b |
| 13 | 5′UCUCAAU|AACUCU3′ | 2.5 ± 0.1 |
| 14 | 5′UUUUUU|UUUUUUU3′ | 0.9 ± 0.1 |
| 15 | 5′UpU3′ | 2.2 ± 0.1 |
Compounds 1–13 are heterooligomers, where the base sequence or the position of the scissile bond has been slightly varied. Compounds 14, a 13-mer uracil homooligomer, and 15, uridylyl-3′,5′-uridine (UpU) have been used as models of a fully flexible phosphodiester bond20 since stacking interactions between uracil bases are known to be of minor importance.35–37 Compounds 1 and 5–7 exhibit faster cleavage than the reference compounds, 8–10 and 13 are equally reactive and the rest of the compounds are cleaved more slowly, so that 2, 11 and 12 are hydrolytically very stable (Table 1).
For all the compounds, MD simulations were performed for structures modelling the TS of phosphodiester bond cleavage within linear single-stranded oligonucleotides. For compounds 4, 9–10 and 14 MD simulations were also run for the initial state structures, whereas for the other compounds, the initial state structures had previously been investigated26 and those MD trajectories were used to analyse the base–base interactions within the flanking sequences of the cleavage site (see below). The features of the initial state structures of compounds 4, 9–10 and 14 are not described here in detail, since they are similar to those reported earlier.26
The general features of the TS structures of oligonucleotides studied
MD simulations of 3.0 ns were performed for compounds 1–14, whose structures represent the TS of phosphodiester bond cleavage within linear single-stranded oligonucleotides. The simulations of 3.0 ns can be considered to be sufficiently long since the continuation of simulations up to 5.0 ns with some compounds did not bring any substantial change in the structures. The stability of structures during the MD simulations was examined by calculating the free energy (G) using the Molecular Mechanics-Poisson-Boltzmann Surface Area (MM-PBSA) method38 as done previously for the initial state structures of the same compounds.26 The G values, the standard errors of the mean and the differences in the free energies between 450–1750 ps and 1750–3000 ps (ΔG) are depicted in Table 2. Both the standard errors of the mean and the ΔG values are very small indicating that the structures are energetically stable during the MD simulations. The average structures of the transition states of oligonucleotides 2 and 6 are shown in Fig. 1. The TS moiety forms a hinge where the flanking sequences are attached. The overall shape of the TS structures of oligonucleotides varies between a curved and extended one. Interestingly, the replacement of purine with uracil seems to straighten the oligonucleotide chain, as seen when the structures shown in Fig. 1 are compared: compound 6, having uracil bases at positions 6 and 8, possesses a more extended structure than 2, having adenosines at these positions.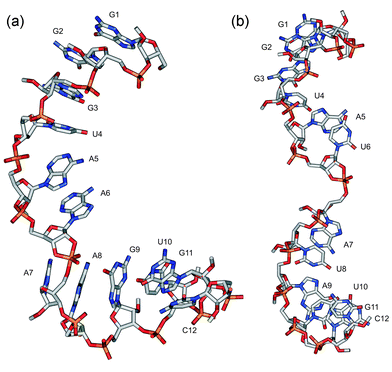 | ||
| Fig. 1 The average structures of the transition states of compounds 2 (a) and 6 (b) during the last 100 ps of molecular dynamics simulations. | ||
| Compound | G(mean)/kJ mol−1a | G(σ)/kJ mol−1b | ΔG/kJ mol−1c |
|---|---|---|---|
| a Mean energies from 680 structures (450–3000 ps).b Standard errors of the mean energies.c G(450–1750ps)-G(1750–3000ps). | |||
| 1 | −11171.4 | 3.0 | 9.6 |
| 2 | −11264.6 | 2.4 | −1.2 |
| 3 | −11724.3 | 2.3 | 7.9 |
| 4 | −11453.0 | 2.7 | 34.7 |
| 5 | −11110.9 | 2.5 | −7.6 |
| 6 | −11127.3 | 2.1 | 3.7 |
| 7 | −10914.6 | 2.8 | 8.2 |
| 8 | −10953.5 | 2.3 | −27.2 |
| 9 | −11350.0 | 3.7 | −23.2 |
| 10 | −11739.4 | 2.8 | −7.1 |
| 11 | −11201.4 | 2.5 | −3.3 |
| 12 | −13412.4 | 3.0 | 27.7 |
| 13 | −11764.9 | 2.5 | −3.6 |
| 14 | −9808.8 | 2.6 | 14.1 |
The structure of the TS moiety
The geometry of the TS moiety was calculated quantum mechanically by optimising the model compound (Fig. 2) at the HF/6-31+G* level first in vacuo and then using a continuum solvation model (water, ε = 80). The geometry obtained is a trigonal bipyramid having two apical (representing 2′O and 5′O atoms) and three equatorial ligands, so that the negatively charged non-bridging oxyligands are in equatorial orientations. The bond angles did not change significantly during the solution optimisation but small changes were observed in bond lengths; the length of the exocylic P–O bond was decreased from 1.89 Å to 1.79 Å and that of the equatorial endocyclic 3′O–P bond from 1.73 Å to 1.66 Å. The trigonal bipyramidal geometry of the TS moiety and the shorter bond lengths of the equatorial bonds compared to the axial ones are consistent with Westheimer's well known concept on pseudorotating oxyphosphoranes.9 The bond lengths and angles obtained from the QM optimisation in solution were used as equilibrium values for the corresponding parameters in MD simulations. The use of equal equilibrium bond lengths for both 2′O–P and 5′O–P can be thought to provide a reasonable model for the TS to be used in MD simulations, although it has been suggested6 that in the hydroxide ion catalysed cleavage of phosphodiester bonds, the rupture of the P–5′O bond is rather advanced.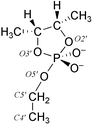 | ||
| Fig. 2 The structure of the model compound used in the quantum mechanical geometry optimisation of the pentacoordinated transition state moiety. | ||
The average structures of the cleavage sites of compounds 2 and 6 in the TS during the last 100 ps of MD simulations are shown in Fig. 3. The conformation of the 3′-linked sugar moiety is C2′-endo. This conformation has been suggested to be39 the most favourable for the in-line displacement. The alignment of the neighbouring bases with respect to the pentacoordinated phosphorane (Fig. 3) depends on the orientations of the flanking sequences, as discussed above (Fig. 1).
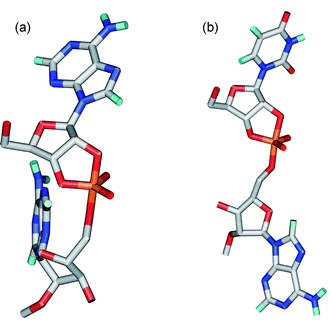 | ||
| Fig. 3 The average structures of the cleavage sites of compounds 2 (a) and 6 (b) in the TS during the last 100 ps of molecular dynamics simulations. | ||
The interactions of pentacoordinated phosphorane moiety
The enhanced reactivity of some phosphodiester bonds within linear single-stranded oligonucleotides compared to a fully flexible phosphodiester bond has been frequently suggested to be due to the TS stabilisation by hydrogen bonding.20,24,28 Especially, direct or water mediated hydrogen bonds from the 5′-linked base to the phosphorane non-bridging oxyligands or to the leaving oxygen atom has been proposed to facilitate the attack on phosphorus and the proton transfer to the leaving group, respectively.20,24,28 Therefore, the distances between possible hydrogen bond donors (the hydrogens of 6-amino group at adenosine) and acceptors (non-bridging phosphorane oxygens (OP) and the 5′-oxygen) at the side of the 5′-linked nucleoside were examined (Fig. 4a). With all the compounds, these distances are longer than 6 Å indicating that direct hydrogen bonding between the base moiety and the phosphate group is not possible. This distance is also too long for the existence of water mediated hydrogen bonds. Within the 13-mer uracil homooligomer, the distances between the phosphorane oxyligands and the carbonyl group oxygen (O4) at the 5′-linked uracil base are longer than 8 Å and definitely too long for water mediated hydrogen bonds. The distance from N7 of the 5′-linked adenine to one of the non-bridging oxyanions, as well as to the leaving 5′-oxygen (Fig. 4b), the protonation of which has been proposed to be particularly important,40,41 in turn, varies between 4.5 Å and 5.0 Å. This suggests the possibility for the water mediated hydrogen bonding. The existence of hydrogen bonds mediated by solvent molecules was extensively examined by determining the distance from one of the non-bridging oxyanions or the leaving 5′-oxygen to the hydrogen atom of a water molecule and the distance from the hydrogen atom of the same water molecule to the N7 of 5′-linked adenine (Fig. 4b). As a criterion for hydrogen bonding a cutoff distance of ≤ 2.5 Å between oxygen/nitrogen and hydrogen atoms were used. The existence of the water mediated hydrogen bond between the non-bridging oxyanions and N7 atom varies slightly between oligonucleotides. Within 6 and 7 having extended 3′-flanking sequences, no water mediated hydrogen bonds were detected, whereas within the other compounds this kind of hydrogen bond was found in 15–30% of all snapshot structures along the MD trajectories obtained (Fig. 4b). Between the leaving 5′-oxyanion and N7, no water mediated hydrogen bonds were detected within any sequences. The distance between O2′ and O2 of 3′-linked pyrimidine bases (Fig. 4a) is ∼ 4 Å. Therefore, surrounding water molecules definitely form hydrogen bonds between these atoms. With 3′-linked purines, this kind of water mediated hydrogen bond is not possible. Although water mediated hydrogen bonds could not be detected in all the compounds, the cleavage site is always well hydrated in the TS regardless of the base sequence. Within a 4.0 Å watershell, there are on average 6.9 water molecules around non-bridging oxyanions (Table 3) and 6.3 water molecules around the leaving 5′-oxygen. The oxyanions tend to form on average 2.8 hydrogen bonds with the solvent molecules and the 5′O on average 1.4 hydrogen bonds (Table 3, as a criterion for hydrogen bonding a cutoff distance of ≤3.5 Å between heavy atoms and a cutoff angle of 90° were used). The whole cleavage site, including the flanking nucleosides is surrounded on average by 70 water molecules (within a 4.5 Å solvation shell). No significant differences between sequences were found neither in the amount of surrounding water molecules nor in the amount of possible hydrogen bonds with solvent molecules.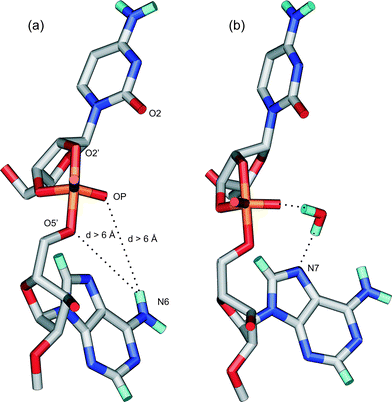 | ||
| Fig. 4 (a) The distances from the non-bridging oxyanions (OP) or the leaving 5′-oxygen to the hydrogens of N6 at the 5′-linked adenosine. (b) The water mediated hydrogen bond, which was found between the non-bridging oxyanion and the 5′-linked adenosine in all other compounds except 6, 7 and 14. | ||
The stacking of bases within the flanking sequences of the cleavage site
The conformational stability of a linear single-stranded oligonucleotide chain is mainly determined by the vertical interactions between bases, i.e. base stacking phenomenon. In order to study the contribution of base–base interactions to the stability of TS within single-stranded RNA molecules, the molecular mechanical (MM) interaction energies between four flanking bases at the cleavage site were calculated both in the initial state and the TS. MM interaction energies were calculated with the parm99 parameter set42 of AMBER 7.0.43 It has earlier been observed that interaction energies calculated with this parameter set correlate well with those calculated quantum mechanically (at the MP2/6-31G*(0.25) level), and accordingly MM interaction energies obtained here can be assumed to provide a correct description of base stacking phenomenon.26,44,45Interaction energies (Eint) between the four flanking bases at the cleavage site (two bases at both sides) in both initial and transition states and the differences in the interaction energies between these two states (ΔEint) for all the compounds studied are recorded in Table 4. In Table 5, interaction energies between two 5′- and two 3′-bases next to the cleavage site (Eint(5′) and Eint(3′), respectively) for both the initial and transition states are shown separately, as well as the differences in the corresponding interaction energies between the initial and transition states (ΔEint(5′) and ΔEint(3′)). As can be seen from Table 4, with heterooligomers 1–13, the stacking at the flanking bases of the cleavage site is enhanced in the TS compared to the initial state, whereas within uracil homooligomer 14, no substantial change in the stacking propensity can be observed on going from the initial state to the TS. In addition, the stacking seems to be, more enhanced within the 5′-linked sequence than within the 3′-linked one (Table 5), although the magnitude of the enhancement is strongly context dependent.
| Compound | Sequence | E int (IS)/kJ mol−1a | E int (TS)/kJ mol−1b | ΔEint/kJ mol−1c |
|---|---|---|---|---|
| The interaction energies are calculated between the underlined bases. a The mean of 36 structures (450–2025 ps). b The mean of 58 structures (450–3000 ps). c Eint(TS) − Eint(IS). | ||||
| 1 | 5′GGGU![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) ![[U with combining low line]](https://www.rsc.org/images/entities/b_char_0055_0332.gif) |GUGC3′ |GUGC3′ |
−39.7 | −51.6 | −11.9 |
| 2 | 5′GGGU![[A with combining low line]](https://www.rsc.org/images/entities/b_char_0041_0332.gif) |GUGC3′ |GUGC3′ |
−43.6 | −47.2 | −3.6 |
| 3 | 5′GGGU![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) |GUGC3′ |GUGC3′ |
−43.6 | −47.3 | −3.7 |
| 4 | 5′GGGU![[G with combining low line]](https://www.rsc.org/images/entities/b_char_0047_0332.gif) |GUGC3′ |GUGC3′ |
−45.7 | −49.3 | −3.6 |
| 5 | 5′GGGU![[U with combining low line]](https://www.rsc.org/images/entities/char_0055_0332.gif) |GUGC3′ |GUGC3′ |
−36.7 | −40.9 | −4.2 |
| 6 | 5′GGGU|GUGC3′ |
−43.9 | −46.9 | −3.0 |
| 7 | 5′GGGU|GUUC3′ |
−43.1 | −53.0 | −9.9 |
| 8 | 5′GUGU|GUGC3′ |
−46.8 | −49.9 | −3.1 |
| 9 | 5′GUGU|GUGC3′ |
−44.2 | −45.2 | −1.0 |
| 10 | 5′GG![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) |AAGUGC3′ |AAGUGC3′ |
−44.7 | −47.4 | −2.7 |
| 11 | 5′GGGUA|UGC3′ |
−42.0 | −47.4 | −5.4 |
| 12 | 5′CCCCA|CCCC3′ |
−44.5 | −46.7 | −2.2 |
| 13 | 5′UCUCA|CUCU3′ |
−48.1 | −49.9 | −1.8 |
| 14 | 5′UUUU|UUUUU3′ |
−26.0 | −26.4 | −0.4 |
| Compound | Sequence | E int(IS, 5′)/kJ mol−1a | E int(IS, 3′)/kJ mol−1b | E int(TS, 5′)/kJ mol−1c | E int(TS, 3′)/kJ mol−1d | ΔEint (5′)/kJ mol−1e | ΔEint (3′)/kJ mol−1f |
|---|---|---|---|---|---|---|---|
| The interaction energies are calculated between the underlined bases. a The interaction energy of two bases at the 5′-side of the cleavage site in the initial state. b The interaction energy of two bases at the 3′-side of the cleavage site in the initial state. c The interaction energy of two bases at the 5′-side of the cleavage site in the transition state. d The interaction energy of two bases at the 3′-side of the cleavage site in the transition state. e E int(TS, 5′) − Eint(IS, 5′). f E int(TS, 3′) − Eint(IS, 3′). | |||||||
| 1 | 5′GGGU|GUGC3′ |
−20.4 | −19.3 | −26.2 | −25.4 | −5.8 | −6.1 |
| 2 | 5′GGGU|GUGC3′ |
−19.7 | −23.9 | −21.7 | −25.5 | −2.0 | −1.6 |
| 3 | 5′GGGU|GUGC3′ |
−21.8 | −21.9 | −21.7 | −25.6 | 0.0 | −3.7 |
| 4 | 5′GGGU|GUGC3′ |
−22.7 | −23.0 | −23.2 | −26.1 | −0.5 | −3.1 |
| 5 | 5′GGGU|GUGC3′ |
−12.6 | −24.2 | −16.1 | −24.8 | −3.6 | −0.6 |
| 6 | 5′GGGU|GUGC3′ |
−22.3 | −21.6 | −24.2 | −22.7 | −1.9 | −1.1 |
| 7 | 5′GGGU|GUUC3′ |
−22.5 | −20.6 | −28.4 | −24.5 | −5.9 | −4.0 |
| 8 | 5′GUGU|GUGC3′ |
−24.7 | −22.1 | −24.1 | −25.8 | 0.6 | −3.7 |
| 9 | 5′GUGU|GUGC3′ |
−21.6 | −22.6 | −16.9 | −28.3 | 4.6 | −5.7 |
| 10 | 5′GG|AAGUGC3′ |
−22.1 | −22.6 | −21.9 | −25.5 | 0.1 | −2.9 |
| 11 | 5′GGGUA|UGC3′ |
−19.9 | −22.1 | −21.3 | −26.1 | −1.4 | −4.0 |
| 12 | 5′CCCCA|CCCC3′ |
−24.2 | −20.3 | −21.2 | −25.5 | 3.0 | −5.2 |
| 13 | 5′UCUCA|CUCU3′ |
−25.6 | −22.5 | −23.2 | −26.7 | 2.4 | −4.2 |
| 14 | 5′UUUU|UUUUU3′ |
−13.8 | −12.2 | −14.0 | −12.4 | −0.2 | −0.2 |
Oligonucleotides 1–4 all have the same base sequence 5′-GGGUAN|AAGUGC-3′ with the exception of the 3′-linked ribonucleoside of the scissile bond, which is either U(1), A(2), C(3) or G(4). The stacking within the AU–AA region of 1 is enhanced more than that within the AN–AA regions (N = A, C, G) of 2–4 (Table 4). With compounds 1 and 2, the stacking within both 3′- and 5′-flanking sequences is enhanced on going to the TS whereas with 3 and 4, the stacking propensity is increased only within the 5′-linked sequence (Table 5). Compounds 1, 5 and 6 are otherwise similar but A5 of 1 is replaced with U5 and A8 of 1 with U8 in 5 and 6, respectively. The stacking is strengthened less within the UU–AA (5) and AU–AU (6) regions than within the AU–AA region of 1 (Table 4). As seen from Table 5, this results from a small change in the stacking tendency within the 5′-linked sequence. The replacement of G11 of 1 with U11 (7) has only a modest influence on the change in the stacking of bases within AU–AA region during the course of chain cleavage (Table 4). A modification close to the 5′-end of the oligonucleotide, G2 (1) → U2 (8), in turn, decreases the enhancement of stacking within the AU–AA region by 8.8 kJmol−1. This originates mainly from the fact that with 8, the stacking of the 3′-linked sequence is similar both in the initial state and TS (Table 5). Within the AC–AA region of 9, the total stacking of bases is not changed significantly on going to the TS, whereas the AU–AA (8) region within the otherwise similar sequence is slightly more strongly stacked in the TS than the initial state (Table 4). The situation is similar to that with 1 and 3, where the enhancement of stacking within AU–AA (1) is stronger than within AC–AA (3). Compounds 1, 10 and 11 have exactly the same base sequence, but the position of the scissile phosphodiester bond is different: between U6 and A7 in 1, U4 and A5 in 10 and A7 and A8 in 11. In all cases, the overall stacking at the flanking bases of the cleavage site is enhanced in the TS, as seen from Table 4. However, with the short 5′-terminal flanking sequence of 10, the stacking remains virtually constant during the chain cleavage (Table 5). Compounds 12 and 13 have the same intervening CAAU|AAC sequence but the terminal sequences differ, being CCC in 12 and UCU in 13. In both compounds, the AU–AA region is stronger stacked in the TS than the initial state but interestingly, this stabilisation is completely caused by enhanced stacking at the 3′-terminal linked sequences.
Discussion
The stabilisation of the TS structures of linear single-stranded oligonucleotides
In the course of the chain cleavage of linear single-stranded RNA molecules the phosphodiester backbone must be rotated in order to obtain the co-linear arrangement between the attacking 2′-oxygen nucleophile and the 5′-oxygen leaving group.9 This rearrangement destroys the base–base interactions across the cleavage site, but appears, in turn, to enhance the stacking of bases in the vicinity, as far as heterooligomers are concerned (Table 4). Within the uracil homooligomer 14, which has been regarded as a model of a random coil oligonucleotide,20 stacking, however, seems to be equally weak in the initial state and the TS. This is expected, since the stacking propensity of uracil bases is known to be negligible.35–37 With heterooligomers 1–13, the magnitude of stacking enhancement within the flanking bases at the cleavage site seems clearly to depend on the base sequence, showing similar features as earlier observed with the initial state structures of the same heterooligomers.26 Stacking appears to be generally more pronounced within the sequence at the 3′-side of the cleavage site than at the 5′-side. This is consistent with the earlier observation, the modification that weakens stacking at a particular position may enhance stacking within the 5′-linked sequence, even several bases away.26 This phenomenon cannot, however, be generalized as seen e.g. with the 3′-terminal flanking sequence of 5 where stacking increases only sightly on going to the TS. This modest change seems to originate from the cooperative stacking propensity of bases within this sequence in the initial state:26 the UUU sequence just at the 5′-side of the cleavage site destroys the continuous stacking within the oligonucleotide chain in the initial state but, in turn, enhances the stacking of A7 and A8.26 Therefore, it can be thought that the stacking of these bases cannot anymore be enhanced by the cleavage.The rotation of the backbone also changes the hydration pattern around the cleavage site. On going to the TS, the cleavage site opens and another negative charge is formed on the phosphate. Evidently, solvation stabilises the TS considerably, but in all likelihood the stabilisation is not significantly dependent on the base sequence. Unfortunately, the available methods do not allow reliable quantitative estimation of the solvation of one part of the system taking the influence of the rest of the system simultaneously into account. In addition, surrounding solvent molecules form hydrogen bonds with both the non-bridging oxyanions and with the leaving 5′-oxygen (Table 3). The tendency to form hydrogen bonds is similar in all the compounds regardless of the base sequence. In addition, apart from 6, 7 and 14, which all have an extended 3′-sequence, water molecule mediated hydrogen bonds from the non-bridging oxyanions to the base of the 5′-linked nucleoside occur (Fig. 4b), but partial protonation of the phosphorane non-bridging oxygens is not a potential source of rate acceleration. By contrast, water mediated hydrogen bonds between the 5′-leaving group and the 5′-linked flanking sequence were not observed. This is most obviously due to the fact that 5′-oxygen is buried by 5′-linked residues. Accordingly, no clearcut evidence for marked sequence dependent stabilisation of the TS by hydrogen bonding could be obtained.
The cleavage of phosphodiester bonds within linear single-stranded oligonucleotides
The cleavage of phosphodiester bonds within linear single-stranded RNA molecules is as a whole a complex problem. The observed reactivity depends on the free energy difference between the initial and TS, and accordingly the sequence-dependent stabilisation of both states affects the cleavage rate. It was recently observed26 that strong base–base interactions across the cleavage site in the initial state correlate with the experimental cleavage rates: strong stacking retards the cleavage. The correlation was based on nine compounds (1–3, 5–8, 12 and 13).26 In order to further investigate the relationship between the reactivity and base–base interactions in the initial state, the interaction energies were also calculated for the initial state structures of compounds 4, 9–11 and 14. In Fig. 5, the interaction energies between four surrounding bases around the cleavage site (the interaction across the cleavage site is also taken into account) for oligonucleotides 1–13 are shown as a function of the logarithm of the rate constant. Of the compounds studied here, 9 and 11 fit very well into the correlation based on the compounds studied earlier,26 whereas 4 and 10 deviate significantly from the others but these deviations can be considered to be in the right direction. Compound 4 is very strongly stacked and unreactive, consistent with the theory that strong stacking retards the cleavage. It should also be noted that 4 is the only compound which has guanosine close to a cleavage site. Within 10, the scissile bond is close to the 5′-end, whereas with all the other compounds, the cleavage takes place in the middle of sequence. Due to this terminal position of the cleavage site, it is expected, that the surrounding bases of the cleavage site within 10 are weakly stacked because they are not stabilised by the 5′-neighbouring bases, and this is observed as an equal cleavage rate with the unconstrained linkage of 14. Stacking within the oligo(U), 14, used as the reference compound, is very weak (E = −32 kJ mol−1, not included in Fig. 5) consistent with the known negligible stacking tendency between uracil bases.35–37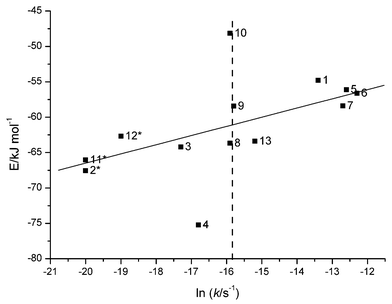 | ||
| Fig. 5 The interaction energies between the four flanking bases of the scissile phosphodiester bond in the initial state structures as a function of the natural logarithm of the rate constant.20 The interaction energies of 1–3, 5–8, 12 and 13 are taken from reference 26. Compounds 2, 11 and 12 are marked with an asterisk because only the upper bounds of the rate constants are known and the cleavage rate of the model compounds of the fully flexible phosphodiester bond is marked with a dashed line (see Table 1). | ||
As mentioned earlier, in addition to the initial state rigidity, the TS stabilisation also plays an important role in the reactivity. This is seen particularly with compounds 1, 5–7. In the initial state, the bases across the cleavage site with these compounds are stacked more strongly than those within the fully flexible polyuridylic acid having negligible stacking, but in spite of that, they are cleaved faster than the reference compound. Accordingly, the TS of compounds 1, 5–7 must be more strongly stabilised than that of the reference compound. As discussed above, solvation, hydrogen bonding with solvent, and base stacking may all contribute to the TS stabilisation. According to the results obtained, hydrogen bonding with water molecules does not appear to result in a sequence-dependent stabilisation of the TS of very labile compounds. While water mediated hydrogen bonds between the non-bridging oxyanions and 3′-terminal sequence were observed for some compounds, these were not found within the most reactive ones. Hence, these calculations suggest that the role of hydrogen bonds with solvent molecules in the sequence-dependent rate accelerations is modest.
TS stabilisation by stacking, in turn, seems to offer a plausible explanation for the rate accelerations over an unconstrained phosphodiester bond. Compounds 1 and 5–7 are the most reactive of the oligonucleotides studied. Enhanced base stacking stabilises the TS of these compounds compared to their initial states by 3.0–11.9 kJ mol−1. In particular, stabilisation of the TS of 1 (ΔEint = −11.9 kJ mol−1) and 7 (ΔEint = −9.9 kJ mol−1) is considerably stronger than with any of the other compounds studied. The weakest TS stabilisation occurs with compounds 9, 12 and 13, which represent average hydrolytic stability. It should, however, be noted that a moderate stabilisation is also observed with the most stable oligonucleotides. Within these compounds strong base–base interactions in the initial state, however, considerably retard the reaction.26 In other words, increase in the overall stacking on going from the initial state to the TS appears to be a plausible explanation for exceptionally rapid cleavage of some phosphodiester bonds in oligoribonucleotides. Within the reference compound uracil homooligomer and similarly also within dinucleotides (e.g. UpU), stacking is negligible both in the initial and the transition states, and evidently, does not affect reactivity. However, other factors such as hydrogen bonding with solvent molecules as well as solvation both of a cleavage site and bases, neither of which is quantitatively investigated here, also undoubtedly play an important role in the reactivity of phosphodiester bonds within oligomeric RNA molecules.
Conclusions
The enhancement of stacking between bases in the course of the reaction stabilises the TS of phosphodiester bond cleavage within heterooligomers, whereas within uracil homooligomers, having inherently weak stacking tendencies, no change in the strength of stacking was observed on going from the initial state to the TS. Hydrogen bonds between the oxyligands of pentacoordinated phosphorane moieties and solvent water molecules appear to be similar in all the compounds regardless of base sequence. Water mediated hydrogen bonds between the non-bridging oxyligands and the 3′-terminal sequence were detected within some compounds but not with the very labile compounds. Hence, these calculations suggest that TS stabilisation by enhanced base stacking, is an important factor that should be taken into account in the analyses of hydrolytic stability of phosphodiester bonds within linear single-stranded RNA oligomers.Methods
The TS model
The force field parameters for the pentacoordinated dianionic phosphorane TS moiety were generated using GAUSSIAN9846 and JAGUAR47 programs and RESP48–50 module of AMBER7.0.43Ab initio quantum chemical calculations were performed using a structure shown in Fig. 2 as the model of a pentacoordinated dianionic phosphorane TS. The geometry of the TS model was first pre-optimised with the monoanionic form of pentacoordinated phosphorane at the HF/3-21G* and HF/6-31G* levels using GAUSSIAN98,45 because the dianionic form is known to be unstable.8 The torsion angle between atoms representing P, 5′O, 5′C and 4′C (Fig. 2) was forced at 180.0° at both stages so that the model would more closely resemble natural RNA. Using this pre-optimised structure as a starting structure the optimisation was continued with a dianionic phosphorane at the HF/6-31+G* level first in vacuo (GAUSSIAN9846) and then in a continuum solvent model (water, ε =80) using the JAGUAR program.47 In addition to torsion restrain, the distance between atoms representing 2′O and P (Fig. 2) was forced at the value of 1.8 Å obtained from the last optimisation with the monoanionic form. Freezing the distance between the 2′-oxygen and phosphorus was necessary, because the rupture of the endocyclic 2′O–P bond is easier than the exocyclic P–5′O bond, as discussed earlier.8The electrostatic potential of the pentacoordinated dianionic phosphorane TS moiety was obtained from ab initio quantum mechanical single-point energy calculations performed for the structure obtained from the solution optimisation. Atom-centered point charges for corresponding atoms were generated from electrostatic potential using RESP methodology.48–50 The charges of the non-bridging oxygen atoms were fixed to be equal as well as those of 5′ hydrogen atoms.
The starting structures
The starting structures for MD simulations were created using the BIOPOLYMER module of the Sybyl program.51 Compounds were initially built up as A-form double-helixes and the complementary strand was then deleted giving the single-stranded oligoribonucleotide with the desired base sequence, and this was used as a starting structure of initial state compounds, as done previously.26 The starting structures for oligonucleotides modelling the TS were constructed rotating manually the backbone torsion angles α, β, γ, ε, and ζ of the cleavage site so that the co-linear orientation between 2′O, P and 5′O would smoothly fit into a single-stranded oligonucleotide helix. In addition, the conformation of the 3′-linked sugar moiety of the cleavage site was changed from C3′-endo, in the A-form RNA, to C2′-endo, which is observed in NMR analysis of internucleosidic linkages inherently in the in-line conformation.39 The 2′-O-methylribonucleoside units were generated in the LEAP module52 of AMBER 7.043 as previously.26 The force field parameters were taken from the parm99 parameter set42 of AMBER 7.043 augmented with parameters for the pentacoordinated dianonic phosphorane moiety. The chimeric ribo/2′-O-methylribooligonucleotides were solvated with a box of TIP3P water extending 13.0 Å in all dimensions around the solute using the LEAP module. This led to box sizes of 51–53–69 Å for initial state structures and 43–52–83 Å for TS structures. The number of water molecules ranged from 4519 to 4938 depending on the size of the oligonucleotide. The system was then neutralised by adding the appropriate number of sodium counter ions.Molecular dynamics simulations and trajectory analyses
Energy minimisations and molecular dynamics simulations were performed using the Sander module of AMBER 7.0,43 using the same procedure as previously for the initial state structures of the linear single-stranded oligonucleotides.26 The resulting MD trajectories were analysed using the ptraj, Carnal, ANAL and MM-PBSA38 modules of AMBER 7.0.43 Calculation of the geometrical parameters was performed using the Carnal and ptraj modules. The energy decomposition between the base moieties within the compound was calculated with ANAL using the same procedure as earlier with the initial structures of these same molecules.26 The absolute free energies of the oligonucleotides studied were calculated as earlier26 using the Molecular Mechanics-Poisson-Boltzmann Surface Area (MM-PBSA) method.38Acknowledgements
The Centre for Scientific Computing (CSC, Espoo, Finland) is gratefully acknowledged for access to the computational resources. This work was supported by the Academy of Finland (grant 74097 to M.P).References
- T. R. Cech, A. J. Zaug and P. J. Grabowski, Cell, 1981, 29, 487–496 CrossRef CAS.
- C. Guerrier-Takada, K. Gardiner, T. Marsh, N. Pace and S. Altman, Cell, 1983, 35, 849–857 CrossRef.
- B. N. Trawick, A. Daniher and J. K. Bashkin, Chem. Rev., 1998, 98, 939–960 CrossRef CAS.
- M. Oivanen, S. Kuusela and H. Lönnberg, Chem. Rev., 1998, 98, 961–990 CrossRef CAS.
- S. Mikkola, U. Kaukinen and H. Lönnberg, Cell Biochem. Biophys., 2001, 34, 95–119 Search PubMed.
- S. Mikkola, M. Kosonen and H. Lönnberg, Curr. Org. Chem., 2002, 6, 523–538 Search PubMed.
- P. Perrault and E. V. Anslyn, Angew. Chem., Int. Ed. Engl., 1997, 36, 432–450 CrossRef.
- D. M. Zhou and K. Taira, Chem. Rev., 1998, 98, 991–1026 CrossRef.
- F. H. Westheimer, Acc. Chem. Res., 1968, 1, 70–78 CrossRef CAS.
- J. W. Strorer, T. Uchimaru, K. Tanabe, M. Uebayasi, S. Nishikawa and K. Taira, J. Am. Chem. Soc., 1991, 113, 5216–5219 CrossRef.
- A. Dejaegere, X. L. Liang and M. Karplus, J. Chem. Soc., Faraday Trans., 1994, 90, 1763–1776 RSC.
- A. Dejaegere, C. Lim and M. Karplus, J. Am. Chem. Soc., 1991, 113, 4353–4355 CrossRef CAS.
- T. Uchimaru, K. Tanabe, S. Nishikawa and K. Taira, J. Am. Chem. Soc., 1991, 113, 4351–4353 CrossRef CAS.
- M. Uebayasi, T. Uchimaru, T. Koguma, S. Sawata, T. Shimayama and K. Taira, J. Org. Chem., 1994, 59, 7414–7420 CrossRef CAS.
- C. Lim and M. Karplus, J. Am. Chem. Soc., 1990, 112, 5872–5873 CrossRef CAS.
- A. Yliniemelä, T. Uchimaru, K. Tanabe and K. Taira, J. Am. Chem. Soc., 1993, 115, 3032–3033 CrossRef.
- K. Taira, T. Uchimaru, J. W Strorer, A. Yliniemelä, M. Uebayasi and K. Tanabe, J. Org. Chem., 1993, 58, 3009–3017 CrossRef CAS.
- A. M. Davis, A. D. Hall and A. J. Williams, J. Am. Chem. Soc., 1988, 110, 5105–5108 CrossRef CAS.
- M. Kosonen, E. Yousefi-Salakdeh, R. Strömberg and H. Lönnberg, J. Chem. Soc., Perkin Trans 2, 1997, 2661–2666 RSC.
- U. Kaukinen, S. Lyytikäinen, S. Mikkola and H. Lönnberg, Nucleic Acids Res., 2002, 30, 468–474 CrossRef CAS.
- U. Kaukinen, L. Bielecki, S. Mikkola, R. W. Adamiak and H. Lönnberg, J. Chem. Soc., Perkin Trans 2., 2001, 1024–1031 RSC.
- B. G. Lane and G. C. Butler, Biochem. Biophys. Acta, 1959, 33, 281–283 Search PubMed.
- K. C. Smith and F. W. Allen, J. Am. Chem. Soc., 1953, 75, 2131–2133 CrossRef CAS.
- R. Kierzek, Nucleic Acids Res., 1992, 20, 5073–5077 CAS.
- R. Kierzek, Nucleic Acids Res., 1992, 20, 5079–5084 CAS.
- U. Kaukinen, T. Venäläinen, H. Lönnberg and M. Peräkylä, Org. Biomol. Chem., 2003, 1, 2439–2447 RSC.
- G. A. Soukup and R. R. Breaker, RNA, 1999, 5, 1308–1325 CrossRef CAS.
- A. Bibillo, M. Figlerowicz, K. Ziomek and R. Kierzek, Nucleosides, Nucleotides Nucleic Acids, 2000, 19, 977–994 Search PubMed.
- R. Stowasser and D. A. Usher, Bioorg. Chem., 2002, 30, 420–430 CrossRef CAS.
- M. Boero, K. Terakura and M. Tateno, J. Am. Chem. Soc, 2001, 30, 8949–8957.
- A. Lahiri and L. Nilsson, J. Mol. Struct., 1997, 419, 51–55 CrossRef CAS.
- D. B. DuPré, I. Vorobyov and M. C. Yappert, J. Mol. Struct., 2001, 544, 91–109 CAS.
- T. M. Popenda, E. Biala, J. Milecki and R. W. Adamiak, Nucleic Acids Res., 1997, 25, 4589–4598 CrossRef CAS.
- P. Auffinger and E. Westhof, Angew. Chem., Int. Ed., 2001, 40, 4648–4650 CrossRef CAS.
- E. G. Richards, C. P. Flessel and J. R. Fresco, Biopolymers, 1963, 1, 431–446 CAS.
- L. D. Inners and G. Felsenfeld, J. Mol. Biol., 1970, 87, 817–833.
- J. Nordberg and L. Nilsson, J. Am. Chem. Soc., 1995, 117, 10832–10840 CrossRef CAS.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case and T. E. Cheatham III, Acc. Chem. Res., 2000, 33, 889–897 CrossRef CAS.
- M. Sassanfar and J. W. Szostak, Nature, 1993, 364, 550–553 CrossRef CAS.
- M. Kosonen, R. Seppänen, O. Wichmann and H. Lönnberg, J. Chem. Soc., Perkin Trans. 2., 1999, 2433–2439 RSC.
- M. Kosonen, K. Hakala and H. Lönnberg, J. Chem. Soc., Perkin Trans. 2., 1998, 663–670 RSC.
- J. Wang, P. Cieplak and P. A. Kollman, J. Comp. Chem., 2000, 21, 1049–1074 Search PubMed.
- D. A. Case, D. A. Pearlman, J. W. Caldwell, T. E. Cheatham III, J. Wang, W. S. Ross, C. L. Simmerling, T. A. Darden, K. M. Merz, R. V. Stanton, A. L. Cheng, J. J. Vincent, M. Crowley, V. Tsui, H. Gohlke, R. J. Radmer, Y. Duan, J. Pitera, I. Massova, G. L. Seibel, U. C. Singh, P. K. Weiner and P. A. Kollman, AMBER 7, 2002, University of California, San Francisco, USA.
- P. Hobza and J. Šponer, Chem.Rev., 1999, 99, 3247–3276 CrossRef CAS.
- J. Šponer, J. Florián, H-L. Ng, J. E. Šponer and N. Špačková, Nucleic Acids Res., 2000, 28, 4893–4902 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 98, Revision A.1, 1998, Gaussian, Inc., Pittsburgh, PA, USA Search PubMed.
- JAGUAR 4.1, 1991–2001, Schrödinger Inc., Portland, OR, USA Search PubMed.
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- W. D. Cornell, P. Cieplak, C . I. Bayly and P. A. Kollman, J. Am. Chem. Soc., 1993, 115, 9620–9631 CrossRef CAS.
- P. Cieplak, W. D. Cornell, C. Bayly and P. A. Kollman, J. Comp. Chem., 1995, 16, 1357–1377 Search PubMed.
- SYBYL, Tripos Associates, 1998, St. Louis, Missouri, USA.
- C. E. A. F. Schafmeister , W. S. Ross and V. Romanovski, LeaP, 1995, University of California, San Francisco, USA.
| This journal is © The Royal Society of Chemistry 2004 |