DOI:
10.1039/B311496C
(Perspective)
Org. Biomol. Chem., 2004,
2, 8-23
Ru complexes bearing bidentate carbenes: from innocent curiosity to uniquely effective catalysts for olefin metathesis
Received
18th September 2003
First published on 24th November 2003
Abstract
The discovery and development of a new class of Ru-based catalysts for olefin metathesis is described. These catalysts, particularly those that do not bear a phosphine ligand, have been demonstrated to promote unique levels of reactivity in a variety of olefin metathesis reactions. The design and development of supported and chiral optically pure variants of this class of Ru catalysts for use in enantioselective metathesis are discussed as well. All catalysts are air stable, reusable, and can be employed with unpurified solvents.
1 Introduction
Since the early nineties and the discovery of structurally well-defined catalysts for alkene metathesis by Schrock and Grubbs, the field of organic synthesis has undergone an exciting transformation.1 Through catalytic olefin metathesis, chemists can now efficiently synthesize an impressive range of molecules that only a decade ago required significantly longer and tedious routes. The primary reason for the success of olefin metathesis is the development of increasingly efficient and selective catalysts.
In this perspective article, we outline the development of a class of Ru carbenes represented by 1–3, that are emerging as increasingly popular metathesis catalysts as a result of their unique properties; three representative transformations that are most effectively promoted by this class of Ru complexes are depicted in Scheme 1
(see below for additional details). The story begins with the serendipitous discovery of Ru-based complex 1 through a set of experiments intended to shed light on the mechanism of various metal-catalyzed ring-opening/ring-closing metathesis (ROM/RCM) reactions.2 Subsequent studies resulted in the availability of non-phosphine Ru carbene 2 and its chiral analogue 3 as catalysts that are recyclable, stable to air or moisture, and operate in the presence of a wide range of common organic functionalities.
 |
| | Scheme 1 Ru-based complexes bearing a bidentate styrene ether ligand serve as effective and practical olefin metathesis catalysts. The reactions shown can only be catalyzed or promoted with high efficiency by this class of Ru complexes. Mes = 2,4,6-Me3C6H2. | |
2 Styrenyl ether Ru complexes: discovery, synthesis and characterization
2.1 Serendipitous discovery of Ru complex 1
In the mid-nineties, one program of research in our laboratories focused on the development of a metal-catalyzed process for efficient conversion of styrenyl cycloalkenyl ethers to 2-substituted chromenes (Scheme 2);2,3 these reactions are promoted by 4–10 mol%
(PCy3)2Cl2Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(H)Ph4
(4) or the more active Mo(CHCMe2Ph)(N(2,6-(i-Pr)2C6H3)(OCMe(CF3)2)2.5 The catalytic metathesis rearrangement, which proceeds through a tandem ROM–RCM, was later used, together with a Zr-catalyzed kinetic resolution, to synthesize optically pure 2-substituted chromenes and the antihypertensive agent (S,R,R,R)-nebivolol (Scheme 1).6
C(H)Ph4
(4) or the more active Mo(CHCMe2Ph)(N(2,6-(i-Pr)2C6H3)(OCMe(CF3)2)2.5 The catalytic metathesis rearrangement, which proceeds through a tandem ROM–RCM, was later used, together with a Zr-catalyzed kinetic resolution, to synthesize optically pure 2-substituted chromenes and the antihypertensive agent (S,R,R,R)-nebivolol (Scheme 1).6
While studying the mechanism of catalytic conversion of styrenyl ethers to chromenes, we found that various metathesis reactions, such as ring-opening metathesis polymerization (ROMP) of 6, are promoted less effectively by 4 when styrene ether 5 is present in solution (Scheme 3). To explain these observations, we proposed that Ru-chelate 1 is formed in situ, and that this complex is catalytically less active (relative to 4) when in the presence of excess styrenyl terminal olefin (e.g., 5).7 We suggested that following the formation of 1
(through metathesis involving styrene 5), the adjacent ether oxygen may associate with the transition metal, to reduce the rate of subsequent propagation steps.
 |
| | Scheme 3 Studies in connection to the mechanism of Ru-catalyzed styrene ethers led to the synthesis and isolation of complex 1. | |
2.2 Synthesis and characterization of Ru complex 1
To gain support for our proposal and the intermediacy of 1 in reactions such as that in Scheme 3, we synthesized, isolated and characterized this metal carbene.8 We showed that when 2-isopropoxystyrene 5 is treated with one equiv. of 4
(24 h), Ru carbene 1 is formed in 67% yield after silica gel chromatography (Scheme 3). As the method of purification suggests, 1 proved to be exceptionally robust.
To avoid the use of stoichiometric amounts of 4 in preparing 1, we developed a two step single-vessel alternative procedure (Scheme 4).8 We established that exposure of Cl2Ru(PPh3)3 to aryldiazomethane 7 results in the formation of a monophosphine 8 without generation of the derived bisphosphine; it is worthy of note that efficient synthesis of complex 8 later proved critical in our ability to synthesize related chiral Ru complexes (cf. Scheme 18). Large, needle-like crystals of 8 were obtained by recrystallization (X-ray structure in Scheme 4). Intermediate 8 need not be isolated; Ru-carbene 1 can be accessed in similar yield when PCy3 is added shortly after exposure of 7 to Cl2Ru(PPh3)3.
 |
| | Scheme 4 One-vessel synthesis of Ru-based metathesis catalyst 1. | |
A range of data support the proposed structure for Ru carbene 1.8 Internal Ru–oxygen chelation is evident in its 1H NMR and 13C NMR spectra. Shielding of the carbene proton (δHα
= 17.44 ppm) results in an upfield shift of ∼2.5 ppm relative to the parent complex (4). The carbene carbon atom resonates upfield (δCα
= 280.63 ppm) in comparison to that of 4
(δCα
= 294.72 ppm). Whereas coupling between the phosphorus nucleus and the carbene proton is absent in 4
(P–Ru–Cα–Hα dihedral angle = 90°), JPH
= 4.4 Hz in the case of 1, suggesting that formation of the five-membered chelate is coincident with a 90° rotation about the carbon–metal double bond. The proposed structure for 1 was further confirmed by single crystal X-ray analysis of naphthyl derivative 9
(Fig. 1). Consistent with the 1H NMR analysis, the Cα–Hα bond of the distorted square pyramidal structure lies in plane with the Ru–P and Ru–O bonds. The Ru–O distance (2.257(7)
Å) in 9 is typical for related O→Ru chelate complexes and suggests that the chelate linkage is reasonably strong.
 |
| | Fig. 1 ORTEP diagram of PCy3Cl2Ru(CH-o-OMeC10H6)
(9). | |
2.3 Synthetic utility of Ru complex 1
Monophosphine Ru carbene 1 promotes ring-closing metathesis (RCM) of five-, six-, seven-, and eight-membered carbo- and heterocycles (Table 1). In each case, the catalyst is recovered chromatographically in high yield as a homogeneous solid residue and maintains its catalytic activity in subsequent reactions.8 As illustrated by the example in Scheme 5, recycled 1 may be carried through at least three additional rounds of RCM. The data presented constitute typical results obtained when recovered residue 1 is transferred to a new reaction vessel followed by the addition of substrate and solvent.
 |
| | Scheme 5 Ru-based metathesis catalyst 1 can be recycled efficiently. | |
| Entry |
Substrate |
Product |
Time/h |
Productb yield (%) |
Rec. catalystb yield (%) |
|
Conditions: 5 mol%
1, CH2Cl2, 22 °C, Ar or N2 atm.
Isolated yields after silica gel chromatography.
Reaction performed in refluxing CH2Cl2.
|
| 1 |

|

|
2.0 |
95 |
89 |
| 2c |

|
|
1.0 |
99 |
88 |
| 3c |

|

|
1.0 |
72 |
95 |
It merits mention that all product isolation work for reactions shown in Table 1 and Scheme 5, including solvent removal following silica gel chromatography, was performed in air with undistilled, reagent-grade solvents; an inert atmosphere is not required to prevent catalyst decomposition. In the solid state, 1 is stable indefinitely in air; in undistilled organic solvents in the presence of water, alcohol, and/or dilute acid (0.01 M HCl), no signs of decomposition (<2%) are evident, according to 1H NMR analysis, after up to one week.8
2.4 Nature of styrene ether and stability of first generation Ru complexes
Ru carbene 10, bearing a methyl ether vs. an i-Pr ether, initially presented itself as an attractive alternative, since the requisite styrene ether can be prepared from an inexpensive and commercially available aldehyde. However, 10 proved to be a less effective catalyst (see example in Fig. 2), and its preparation led to several complications. In a solution of undistilled chloroform in air, 10 slowly decomposes over a period of several weeks to produce o-anisaldehyde (through oxidation of the metal carbene). In the case of 1, after two weeks under identical conditions, there is <2% decomposition. Moreover, unlike 1, complex 10 cannot be recovered in high yield after silica gel chromatography.
 |
| | Fig. 2 Substitution of the Oi-Pr (catalyst 1) group with an OMe group (catalyst 10
) significantly reduces the stability and metathesis activity of the Ru complex. | |
The difference in catalytic activity of 10 and 1 indicated that the nature of the ether chelate is critical to the stability, as well as activity, of a styrene ether Ru complex.8 We suspected that the differences in activity and stability of the methoxy- (10)
vs. isopropoxy (1) Ru-carbene complexes arise from the relative steric bulk of these two substituents. Since catalytic metathesis likely proceeds through a dissociative mechanism (see Scheme 6), we argued that the larger i-Pr group may facilitate dissociation of the oxygen atom from Ru during catalyst initiation. It is also tenable that the isopropoxy group is a more robust ligating unit due to its higher Lewis basicity, offering more effective electronic stability to 1. As will be discussed later, such considerations proved useful in the development of subsequent generations of this class of Ru catalysts (cf. 2c in Fig. 8 and 3d and 3f in Scheme 20).
 |
| | Scheme 6 Proposed route for the release, catalytic activity and return of Ru complex 1. | |
The stability of 1 implies that increased sterics might allow for more effective protection of the metal center from undesirable side reactions (e.g., carbene oxidation). In this context, it has been reported that the stability of bridged-chloride Ru-carbene complexes can be critically dependent on steric requirements of the ligand environment surrounding the metal center.9 Indeed, as illustrated in Fig. 3, Ru complex 11 bearing the smaller OMe group, an intermediate synthesized en route to 10, crystallizes as a dimeric entity containing bridging anionic chloride ligands.
 |
| | Fig. 3 Ru complex 10 bearing styrene methyl ether (vs. Oi-Pr) is less stable and not as catalytically active. The derived PPh3 complex 11 crystallizes as a dimer, underlining the reduced sterics at the metal center. | |
2.5 Mechanistic considerations regarding catalytic activity of complex 1
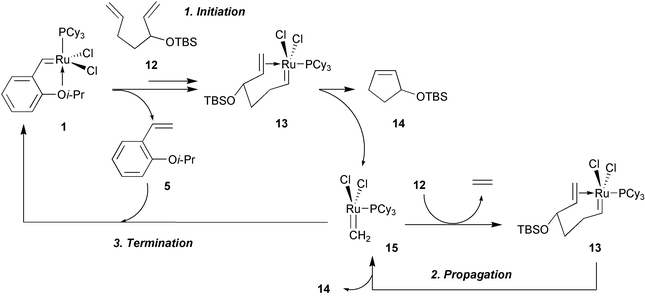
A plausible mechanistic scenario can be proposed that accounts for RCM activity and recyclability of Ru complex 1
(Scheme 6). Formation of carbene 13 results in the release of styrene ether 5. Subsequent conversion of 13 to RCM product 14 through the derived metallacyclobutane intermediate releases monophosphine Ru-methylene 15 to complete the initiation stage. With an excess of the diene substrate present, the highly reactive 15 enters the propagation cycle to promote additional product formation (concomitant with the release of ethylene). Complex 15 may encounter 5, leading to regeneration of 1; as the concentration of 12 decreases, initiation of 1 is also reduced to cause efficient catalyst recovery.10
The influence of internal chelation on the initiation and propagation rates of 1 relative to benzylidene 4 was probed by monitoring the ROMP of cyclooctene (Fig. 4).7 Pseudo first-order rate constants for initiation (consumption of 1) and propagation (formation of 16) were measured by integration of the Hα resonances of carbenes 1 and 16 and the olefinic proton signals of cyclooctene and 16, respectively (1H NMR). These experiments indicated that 1 initiates approximately 30 times slower and propagates nearly four times faster than 4. The slower initiation (1vs. 4) may be due to the less facile dissociation of the smaller isopropyl aryl ether ligand (relative to PCy3) from the sterically congested metal center (see also Scheme 14). In addition, in the case of 1, generation of the active complex (e.g., 13) requires dissociation of the aryl ether ligand as well as a metathesis step (activation of 4 only requires phosphine dissociation). Re-association of the alkoxy unit to the transition metal center should be rate-inhibiting and more favored on entropic grounds (alkoxy styrene is bound to the transition metal at two sites). The enhanced propagation of 1 is consistent with the intermediacy of monophosphine 15
(Scheme 6) and the rate-retarding effects of excess phosphine in Ru-catalyzed metathesis reactions; it is likely that PCy3 is more effective at re-associating with a Ru carbene (slowing rate of metathesis) than ether 5.11
3 Second generation non-phosphine Ru-based olefin metathesis catalyst
3.1 Synthesis and characterization of Ru complex 2
Despite the attractive attributes of 1, this catalyst for the most part proved to be an efficient metathesis catalyst only with substrates that contain terminal alkenes. To address the question of reactivity without altering the structural features that allow the catalyst to be recyclable, in 1999 we synthesized, characterized and examined the catalytic activity of Ru complex 2.12 Our adopted strategy was based on the accelerating effect of a variety of saturated imidazolin-2-ylidene13 and unsaturated14 imidazol-2-ylidene carbene ligands15 on the activity of Ru-based metathesis catalysts. We established that, as depicted in Scheme 7, treatment of Grubbs's second generation catalyst 1712a with 1.0 equiv. CuCl and 0.97 equiv. 5 in CH2Cl2 at 40 °C leads to the formation of 2 within 1 h. Ru complex 2 can be isolated in air as a bright green solid in 85% yield after silica gel chromatography (mp = 178–181 °C dec.). Single crystal X-ray analysis of 2
(Scheme 7) confirmed the structural assignment (see Scheme 2).
 |
| | Scheme 7 First generation synthesis and X-ray structure of non-phosphine Ru-based complex 2. Mes = 2,4,6-Me3C6H2 | |
Comparison of the 1H NMR spectra of 1 and 2 points to their subtle structural characteristics. As illustrated in Fig. 5, there are two distinct chemical shift changes in the 1H NMR spectra of 1 and 2; one variation is observed at the i-Pr methine proton and another at the carbene CH (Hα). In both instances, the protons for the imidazolin-2-ylidene system 2 are more shielded. These differences are likely due to higher electron density at the transition metal center of 2, caused by the stronger electron donation by the heterocyclic ligand (relative to PCy3).12 The weaker electron donation by the oxygen ligand to the Ru center in 2 is manifested by the more upfield appearance of the isopropyl methine proton (4.90 vs. 5.28 ppm). However, the difference in the chemical shifts of Hα may be partially due to an anisotropic effect caused by the aryl units of the heterocyclic ligand in 2.
 |
| | Fig. 5 Selected spectroscopic differences between Ru complexes 1 and 2. | |
3.2 Ru-based complex 2: a recyclable and highly active metathesis catalyst
As the representative data in Table 2 illustrate, Ru complex 2 is a highly effective catalyst for RCM of dienes; trisubstituted (entry 1) and 1,1-disubstituted (entry 2) olefins can be utilized in the synthesis of trisubstituted cyclic alkenes. As indicated by the catalytic RCM in entry 3, trisubstituted allylic alcohols can be accessed in the presence of 5 mol%
2.16 Catalyst loadings lower than 5 mol% are sufficient; as exemplified by the reaction in entry 1, catalytic RCM can readily proceed to completion with only 1 mol%
2. Tetrasubstituted olefins are obtained through catalytic RCM promoted by 5, albeit less efficiently (entries 3–4, Table 2). The lower levels of efficiency observed in the synthesis of tetrasubstituted alkenes may be because the released styrenyl ether effectively competes with 1,1-disubstituted olefins to re-form the initial Ru carbene, thereby diminishing formation of the requisite Ru-carbene derived from the triene substrate. In addition, the catalyst (or the released Ru-methylene) may undergo partial decomposition under metathesis conditions at the required 80 °C for more than a few minutes.
| Entry |
Substrate |
Product |
Time |
Conv (%) |
Product yield (%)b |
Catalyst recovery (%)b |
|
Conditions: 1 mol%
2 for entry 1, 5 mol%
2 for entries 2–4; 22 °C, CH2Cl2 for entries 1–2, 24 h at 22 °C and 20 h at 40 °C, CH2Cl2 for entry 3; toluene, 80 °C for entry 4.
Isolated yields.
|
| 1 |

|
|
20 min |
>98 |
87 |
98 |
| 2 |

|
|
2 h |
>98 |
75 |
95 |
| 3 |

|
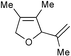
|
44 h |
38 |
81 |
|
| 4 |

|
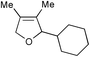
|
30 min |
70 |
65 |
60 |
As was the case with complex 1, catalyst 2 can be recovered with high efficiency after silica gel chromatography (recrystallization not needed) and used in subsequent reactions with equal efficiency. It merits mention that monophosphine Ru catalyst 1 is significantly less efficient in promoting the transformations shown in Table 2. As an example, treatment of the alcohol in entry 2 of Table 2 with 5 mol%
1
(22 °C) leads to only 15% conversion after 2 h.
3.3 Synthetic utility of Ru complex 2: more than just a recyclable alternative
A number of reports from various laboratories have appeared indicating that Ru catalyst2is not only a recyclable metathesis catalyst, but also offers reactivity levels unavailable by the corresponding phosphine-bearing complex17. Complex 2 thus expands the scope of metal-catalyzed olefin metathesis. Representative examples are reviewed below. It must be noted that the point of the discussion below is not to suggest that 2 is superior to 17, but to alert the reader of the advantages that can be offered by Ru complex 2. It should also be noted that 2 and 17 are commercially available (Aldrich).17
3.3.1 Utility in catalytic cross metathesis (CM) reactions.
The earliest examples suggesting that Ru complex 2 may have unique properties as a metathesis catalyst appeared in the context of cross metathesis (CM) reactions involving electron deficient olefin partners. Several examples are shown in Scheme 8
(see Scheme 10 for an application to target-oriented synthesis). The catalytic CM shown in Scheme 8a has been reported by Cossy to occur site-selectively at the more electron-rich homoallylic olefinic site to deliver the α,β-unsaturated aldehyde in 73% isolated yield.18 The reaction of acrylonitrile with a variety of terminal alkenes, such as that illustrated in Scheme 8b, cannot be effected in the presence of phosphine-bearing Ru complex 17.11,19 In contrast, Blechert et al. have disclosed that with 5 mol%
2, CM proceeds readily to afford the desired products, predominantly as their Z isomer, in high yields.20 In the course of investigations in our laboratories in connection to the development of new methods for catalytic asymmetric conjugate additions of alkylzincs to enones, we observed that many of the requisite α,β-unsaturated enones can be easily accessed in >90% isolated yield through effective catalysis by 1–5 mol%
2
(Scheme 8c).21 However, in most such cases, use of catalyst 17 led to the generation of a number of undesired products. The reaction in Scheme 8d is one of several examples reported by Cossy et al. in their report outlining the ability of Ru complex 2 to effect CM of various allylsilanes and unsaturated aldehydes, ketones, esters and carboxylic acids.22 The example depicted in Scheme 8e was recently disclosed by Grimaud and coworkers. Attempted tandem enyne RCM–CM involving 18 and unsaturated ester 19 with 5 mol%
17 leads to the formation of 20 as the major product in 50% yield, the transformation does not venture beyond the initial RCM stage (exclusive generation of 20).23
 |
| | Scheme 8 Non-phosphine Ru complex 2 offers reactivity levels in effecting CM reactions that are not available by related systems such as catalyst 17. | |
Since Ru-based complexes are capable of carrying out transformations other than olefin metathesis,24 an emerging area of investigation involves the development of tandem catalytic protocols.25 In this context, as illustrated in Scheme 9, Cossy and coworkers have shown that Ru catalyst 2 can be used to effect efficient tandem Ru-catalyzed CM–catalytic hydrogenation to access organic molecules which would otherwise have to be prepared by less efficient routes.26 It should be noted that similar tandem transformations in the presence of Ru complex 17 require more forcing conditions.
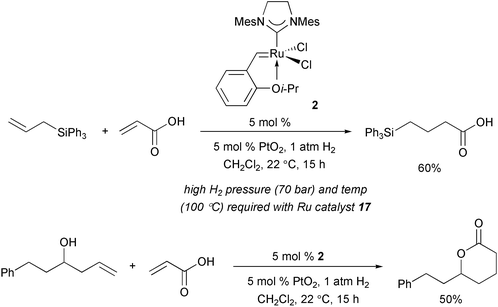 |
| | Scheme 9 Tandem CM–hydrogenation reactions promoted by Ru complex 2 under mild conditions. | |
3.3.2 Utility of Ru complex 2 in synthesis of biologically active molecules.
The unique ability of Ru carbene 2 as an olefin metathesis catalyst has been exploited in a number of studies directed towards syntheses of biologically active molecules. As illustrated in Scheme 10, Cossy et al. have utilized complex 2, in conjunction with Ti-allyl reagent 22, to effect two catalytic CM–allyltitanation sequences and another catalytic CM to develop a stereoselective and efficient synthesis of the C1–C14 segment of amphidinol 3.27
 |
| | Scheme 10 Sequential CM–enantioselective allylation involving Ru catalyst 2 used by Cossy in an efficient synthesis of a segment of amphidinol 3. PMP =
p-methoxyphenyl. | |
In a recent enantio- and stereoselective total synthesis of topoisomerase II inhibitor (R)-(−)-elenic acid, we utilized a sequential Ru-catalyzed homodimerization–hydrogenation in the presence of 5 mol%
2 to convert unsaturated acetate 23 to saturated bis(acetate)
24
(Scheme 11).28 At a later point, CM of 1,1-disubstituted olefin 25 with optically enriched chiral terminal olefin 26 was effected with 35 mol%
2 to afford trisubstituted alkene 27 in 40% isolated yield as a 3 : 1 mixture of E : Z olefin stereoisomers. Related studies indicated that use of Ru complex 17 to effect CM with chiral terminal alkenes such as 26 can lead to ∼10% reduction in optical purity. The high catalyst loading required in the synthesis of 27, as well as the moderate stereoselectivity observed, point to the need for the development of more effective catalysts for this important class of CM reactions.29
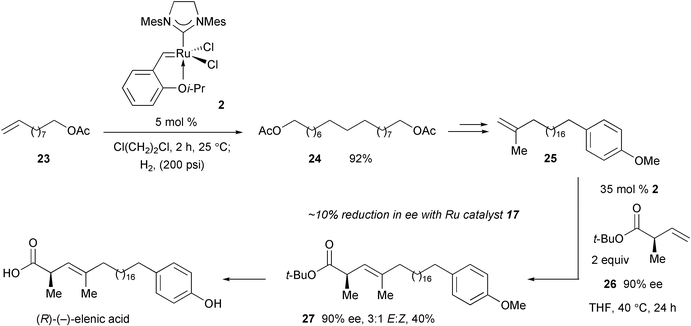 |
| | Scheme 11 Use of sequential CM–hydrogenation and another stereoselective CM catalyzed by Ru complex 2 in the enantioselective total synthesis of (R)-(−)-elenic acid. | |
Another instance where the unique catalytic activity of Ru carbene 2 is underlined is in the preparation of cyclosporin A analogues obtained through catalytic CM of the immunosuppressant with a range of other α,β-unsaturated esters (Scheme 12a). Lazarova et al. report that 2
“proved to be the best catalyst for this cross metathesis” reaction and that the catalytic coupling is a “highly scalable process” and delivers 85–90% yields of >95% pure products.30 Conversion of a crotyl side chain to a desired vinyl group was reported by Wipf and coworkers to be effected in the presence of 2, p-TsOH and ethylene in the context of a total synthesis of the alkaloid (−)-tuberostemonine (Scheme 12b).31 These researchers point out that “phosphine-free conditions were important to avoid extensive chromatographic purification that led to decomposition.” Interestingly, the Ru-catalyzed CM is followed by a catalytic hydrogenation. Assuming that the presence of sulfonic acid does not bear detrimental consequences, it would be intriguing to consider whether a one-pot process involving Ru-catalyzed CM–hydrogenation would be successful in this case.
 |
| | Scheme 12 Additional examples of Ru catalyst 2 serving a unique role in syntheses of biologically active molecules; a: mol% not reported. | |
The final example shown in Scheme 12 involves a sequential Ru-catalyzed enyne RCM–CM that was recently encountered in these laboratories en route to the total synthesis of erogorgiaene.32 Whereas both Ru complexes 2 and 17 efficiently promote the formation of cyclic 1,3-diene 28, it is the non-phosphine complex (2) that promotes the subsequent catalytic CM with methyl vinyl ketone to afford 29
(>98% conv. vs. 30% conv. with 17 after 12 h).
3.3.3 Utility in catalytic ring-closing metathesis (RCM) and applications to syntheses of biologically significant molecules.
The unique attributes of Ru complex 2 have been demonstrated in the context of RCM processes as well. Hale and coworkers report that conversion of highly functionalized diene 30 to cyclopentenyl adduct 31, which is an intermediate in the total synthesis of anticancer (−)-agelastatin A, is best effected in the presence of 2
(Scheme 13).33 In another example, Reiser et al. have recently disclosed that the challenging catalytic RCM of diene 32 to afford polycyclic 33 can only be promoted in the presence of Ru catalyst 2. When phosphine complex 17 was used, no reaction was observed.34
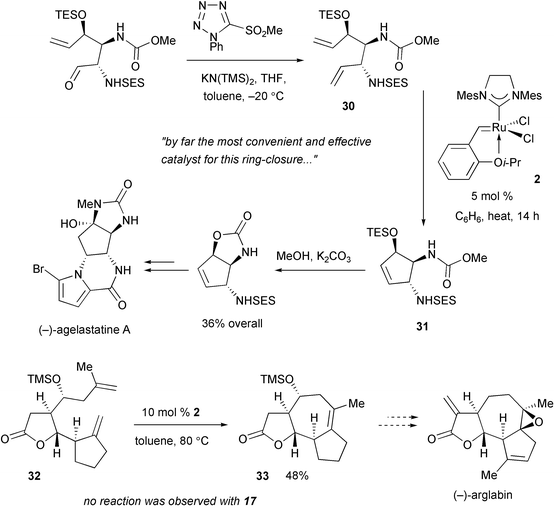 |
| | Scheme 13 Examples of RCM reactions where Ru complex 2 uniquely provides an effective solution. | |
3.3.4 The mechanistic origin of unique catalytic activity of non-phosphine Ru catalyst 2.
The origin of unique activity of Ru complex 2 may be due to the fact that it does not bear a phosphine ligand and thus, in the course of catalytic metathesis, there is no free phosphine in solution. It has been demonstrated that phosphine ligands might suppress catalyst activity in Ru-catalyzed olefin metathesis through competition for open ligation sites in the catalytically active 14-electron intermediate (b in Scheme 14).19 That is, free phosphine in solution can inhibit coordination of olefins to the transition metal center by re-association with the active Ru complex (c→b in Scheme 14). In a similar fashion, with non-phosphine Ru complex 2 activation occurs through loss of O→Ru chelation (2→d, Scheme 14); however, in this case, the styrenyl ether ligand is less efficient at re-binding the active transition metal complex (c in Scheme 14) and, therefore, competes less effectively with olefin substrates for Ru chelation. With Ru complex 2, efficient turnover can occur without sequestration of the active complex b
(see Scheme 14). Such effects are likely to be pronounced in catalytic CM involving electron-deficient alkenes. With electron-withdrawing carbenes (e.g., R = CN in b in Scheme 14), chelation of the Lewis basic PCy3 should be more favored and catalytic activity with 17 can suffer significantly. As validated by experimental data (cf. Schemes 8, 11 and 12), for metathesis reactions involving potential formation of an electron-deficient carbene, a phosphine-free catalyst (e.g., 2) is best suited.35
 |
| | Scheme 14 The absence of a phosphine ligand in Ru catalyst 2 avoids formation of complexes such as c
(formed when 17 is used) which may not be able to re-enter the catalytic cycle readily. | |
Such a mechanistic proposal is supported by previous studies disclosed by Grubbs and coworkers in connection to catalytic metathesis reactions involving difluoroethylene.36 In the above study, it is demonstrated that once an electron deficient difluoromethylidene is formed, it rapidly associates with a phosphine ligand to afford a complex that is reluctant towards reinitiation; only higher temperatures or additives (such as CuCl or HCl to promote phosphine dissociation) re-establish the activity of the difluoromethylene Ru complex.
4 Supported variants of Ru-based metathesis catalysts 1 and 2
4.1 Dendritic complexes
The structural robustness and synthetic utility of Ru complexes 1 and 2 suggest that their supported variants should also be of significant utility.37 With the availability of easy-to-handle, efficient and recyclable supported Ru complexes, catalytic metathesis could be extended to preparative synthetic and combinatorial chemistry.
In 2000, we reported the synthesis and catalytic activity of Ru-based dendrimers 34 and 35.12 We initiated our studies of supported metathesis catalysts with dendritic systems because of their ease of characterization and the high level of certainty with which metal-containing sites can be introduced at their periphery. With a catalyst based on these small branching polymers, it would be possible to gauge rigorously the efficiency with which the active metal carbene leaves the ligation site and returns to the macromolecule (cf. Scheme 6).
The high solubility of dendrimers 34 and 35 in organic solvents permitted full analysis by NMR spectroscopy and high-resolution mass spectrometry. Multi-component catalyst 34 exhibits activity similar to monomeric 1. Furthermore, product isolation is simple: the reaction mixture is passed through a short silica gel column. Subsequent washing of silica gel led to quantitative recovery of the dendritic catalyst. Our studies indicate that, after one representative Ru-catalyzed RCM (see Scheme 15), 13% of the styrene ligands on the dendrimer become vacant – presumably due to Ru complex decomposition (1H NMR analysis). Repeated use of recycled 34, in spite of this steady Ru loss per reaction, promoted facile RCM and the desired product was isolated in >86% yield. The dendritic complex remained active even after 50% of its sites were depleted of Ru (see cycle 6, Scheme 15). This high level of reactivity suggests the intermediacy of a coordinatively unsaturated, monophosphine carbene (15, Scheme 6). As expected, non-phosphine dendrimer 35 exhibited higher levels of activity (see Scheme 14); as an example, the formation of the cyclohexenyl allylic alcohol proved significantly slower in the presence of 34. Attempts to avoid chromatography and recycle the dendrimers 34 and 35 by precipitation in the presence of a variety of solvents proved unsuccessful.
 |
| | Scheme 15 Dendritic variants of Ru complexes 1 and 2 and their activity as olefin metathesis catalysts. | |
The recycling experiments shown in Scheme 15 indicate that substantial turnover can accompany minor amounts of Ru release. At this point, the question arises as to whether any of the released Ru carbene ever returns to the styrene ether site. Our efforts to address this issue were facilitated by a minor chemical shift difference for the carbene proton signals of 1 and 34; this allowed us to determine the amount of Ru bound to dendritic versus monomeric styrene ligands by integration of the appropriate downfield signals in the 1H NMR spectrum of a mixture.12 In a control reaction, as illustrated in Scheme 16, we established that prolonged treatment of 34 with 2-isopropoxystyrene (5) results in <2% metal crossover. However, repetition in the presence of an olefin substrate results in RCM and statistically-driven scrambling of the transition metal between monomeric and dendritic ligation sites within 15 minutes. These results imply that the Ru center, after reacting with a substrate alkene and leaving the dendrimer, can be trapped again by a styrene ether. These findings further suggest that a significant portion of the available metal initiates the moment a metathesis reaction begins, providing direct evidence for the ‘release/return’ mode of action (Scheme 6).
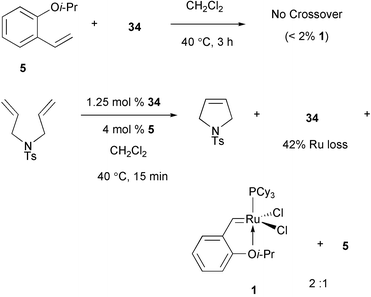 |
| | Scheme 16 Experiments regarding return of Ru carbenes to dendrimer surface. | |
4.2 Polymer-supported variants of Ru complex 1
Two polymer-supported versions of monophosphine Ru complex 1 have been prepared. PEG-supported catalyst 36 has been reported by Yao.38 The choice of PEG as the carrier is noteworthy in Yao's system, as it permits catalysis under standard homogeneous conditions and enables easy recovery of the catalyst by precipitation and filtration. Supported catalyst 36 effectively promotes RCM of terminal olefins, including medium ring structures. Precipitation, filtration, and re-use of the recovered catalyst gives high conversion in a second round of metathesis. In fact, there was little or no loss in activity after eight runs of recycling. All operations, including concentration of the reaction mixture, precipitation, filtration, and washing with reagent-grade diethyl ether can be carried out in air.
Dowden and Savovic39 have disclosed a complementary strategy based on a subtle change in the site of polymer attachment. With Ru complex 1 again serving as a model, these workers manipulated the isopropyl portion of the styrene ether as a covalent linker unit to prepare 37. Polymer-supported catalyst 37 readily promoted RCM reactions of monosubstituted olefins; general laboratory-grade methylene chloride was used without degassing in air. Catalytic metatheses were performed by addition of substrate solutions to the resin in a solid plastic tube fitted with a glass frit which was then sealed and subjected to 360° rotation. Filtration and washing with methylene chloride directly afforded the product and recovered catalyst. Good yields were reported in certain cases when the catalyst was recycled over five runs; however, a decreased catalyst loading of 1.5 mol% resulted in less effective recycling.
More recently, Yao and Zhang have reported a variant of Ru catalyst 1 that bears an ionic tag (38, Fig. 6).40 Complex 38 promotes RCM of dienes in ionic liquid [Bmim]PF6 and CH2Cl2
(1 : 9 v/v) to afford cyclic disubstituted cyclic alkenes. The catalyst can be recycled. In one instance, ten cycles of RCM required an equal length of time (3 h) to generate an unsaturated seven-membered ring amide. Nevertheless, in another case, reactivity is diminished significantly after two cycles. Reactions seem to require elevated temperatures (55 °C) and are run under Ar; moreover, multiple washes with Et2O are used to obtain the product. Such factors detract from this interesting system, particularly when its use in a library synthesis and in large scale preparations are being considered.
 |
| | Fig. 6 Supported variants of Ru catalyst 1. | |
4.3 Polymer-supported variants of Ru complex 2
We have reported a procedure for the surface derivatization of small glass (sol-gel)41 pellets and applied this procedure to the synthesis of supported Ru catalysts (Scheme 17, 39→41).42 Accordingly, treatment of 39 with allylchlorodimethylsilane and a full equivalent of Ru complex 17 led to rapid ROM/CM and metallation of the styrenyl ether (→40). Pre-weighed monolithic (smallest dimension ≥ 1 mm) sol-gels were then added to the solution, and substitution of the labile Si–Cl bond in 40 with free hydroxyl groups on the glass surface anchored the catalyst to the support, affording dark green glass pellets (41). Sol-gel glass was selected for catalyst support for several reasons: (1) These porous glasses retain a rigid and exposed interfacial surface area (typically 300–1000 m2 g−1), whereas organic polymer beads swell and shrink in different solvents, often with unpredictable effects on catalysis. (2) Functionalization of a monolithic (smallest dimension ≥ 1 mm) gel affords a bulk catalyst sample; this obviates the need for filtration to recover the catalyst – tweezers can be used instead. (3) Gelation occurs after a sol is cast into a mold. The glass pieces can therefore be tailored to a uniform size or shape.
Sol-gel-supported Ru complex 41 proved to be a remarkably efficient and recyclable catalyst. With purified solvent under N2, as illustrated in Scheme 17, catalyst 41 was used in the synthesis of a trisubstituted olefin for a total of twenty cycles. Moreover, the glass-bound Ru complexes were used to effect synthesis of two small libraries through catalytic RCM (6 hours at 22 °C under air with reagent-grade methylene chloride). For reactions that proceeded to completion and delivered a single product, representative elemental (C,H) and ICP-MS analyses showed that the products – without purification or workup – were of high (often analytical) purity and the level of Ru contamination was typically <1%.
Synthesis and catalytic activity of supported Ru catalysts 42 and 4343
(Scheme 17) have been reported by Blechert et. al. One significant difference between these two complexes is that with 42, the Ru carbene remains bound to the support, whereas in the case of 43, similar to glass-supported 41, the metal carbene is likely released into solution. Both catalysts effect RCM reactions effectively; re-use up to four cycles is reported, although reaction times have not been provided in all cases. Studies regarding CM revealed notable differences in catalyst efficiencies. As the representative data in Scheme 17 illustrate, complex 43 displays enhanced activity in the reaction of a terminal olefin of a β,γ-unsaturated ester with a variety of electron-deficient partners. Although 42 and 43 are bound to distinct polymer supports, making a direct comparison difficult, the higher activity of 42 is likely a function of its ability to release the active complex. Since the propagating carbene from 43 remains bound to support, diffusion of reacting alkenes into the cavities of the polymer is rate-limiting – especially with the more challenging CM reactions.
 |
| | Scheme 17 Several early examples of supported variants of Ru complex 2 and representative reactivity data. | |
Grela et al. have reported the synthesis and catalytic activity of the polystyrene-supported Ru complex 44
(Fig. 7).44 The catalyst shows high activity, as it readily promotes RCM to afford trisubstituted olefins. Relevant examples involving syntheses of medium and large ring structures are provided; moreover, complex 44 can be recycled, although by the fifth cycle significantly longer reaction times seem to be required. Blechert and coworkers recently disclosed the synthesis and catalytic activity of polymeric Ru-based catalyst 45
(Fig. 7).45 Attractive features of complex 45 include ease of synthesis and low levels of Ru impurity detected in product mixtures, suggesting that small amounts of highly active Ru carbenes are released upon exposure to substrate molecules. However, in spite of the presence of the imidazolinium ligand only reactions of terminal alkenes leading to disubstituted olefins are reported, and in one recycling study (involving formation of a disubstituted cyclic amide), the catalyst loses significant activity after the seventh cycle.
 |
| | Fig. 7 More recent supported variants of Ru complex 2. | |
5 Chiral non-phosphine Ru-based catalysts for enantioselective olefin metathesis
A critical objective in the field of catalytic olefin metathesis relates to the design and development of chiral versions of this class of catalysts that can be utilized in enantioselective olefin metathesis.46 With the availability of such chiral complexes, a variety of optically pure compounds become accessible in an efficient and practical manner. It was within this context that in 2002, we reported the synthesis, structure and reactivity of a chiral non-phosphine Ru carbene 3.47,48
5.1 Synthesis of the first generation chiral Ru complex
Optically pure 3 was synthesized as shown in Scheme 18; all reactions were performed on gram scales. The critical step is the conversion of 46 to 3. After extensive experimentation, we established that in the presence of silver carbonate and the catalytically inactive Ru complex 9, the desired complex 3 is formed in 52% isolated yield. The optically pure non-phosphine complex 3, bearing a stereogenic Ru center, was isolated as a single diastereomer.49 Ru complex 3 is air-stable, can be purified by silica gel chromatography with undistilled solvents and its diastereo- and enantiomeric purity was established by HPLC analysis (isolated in >98% de and ee).
 |
| | Scheme 18 Synthesis and X-ray structure of non-phosphine Ru-based chiral metathesis catalyst 3. | |
5.2 Utility in asymmetric ring-opening/cross metathesis (AROM/CM)
Chiral Ru catalyst 3 promotes asymmetric ring-opening/cross metathesis (AROM/CM)50 in air, with undistilled solvents and with substrates that readily polymerize with chiral Mo catalysts (Scheme 19).51 Complex 3 can be recovered after chromatography (86–71% yield) and can be re-used without significant loss of enantioselectivity and reactivity. The catalytic AROM/CM in Scheme 19 demonstrate the synthetic potential of chiral Ru catalysts. Enantioselective metatheses can be promoted efficiently and enantioselectively, with as low as 0.5 mol% catalyst loading, at room temperature, in air and with undistilled and non-degassed solvent. Even reactions run at 50 °C can be run in air without significant reduction in reactivity or selectivity.
 |
| | Scheme 19 AROM/CM promoted by Ru-based chiral metathesis catalyst 3. | |
5.3 Second generation chiral Ru complexes: higher reactivity and expanded scope
In spite of the promising levels of selectivity observed in catalytic reactions of 3, this chiral catalyst proved to be less reactive than its achiral analogue 2, probably as a result of steric (large chiral ligand) and electronic factors (an aryloxide vs. a Cl group).52 To access more active catalysts, we have most recently prepared several new optically pure Ru carbenes 3a–3f
(Scheme 20) through modifications of the benzylidene and chiral ligands in 3.53 Chiral catalyst 3a, bearing the electron-withdrawing NO2
(para to the ligating Oi-Pr) was investigated based on the expectation that the nitro substituent would weaken i-PrO→Ru chelation and facilitate initiation of the catalytic cycle. A similar influence was expected from the electron-releasing OMe (para to the RuC bond) in 3c, where increased electron donation into the metal center would reduce its Lewis acidity. The above hypotheses were based on reports by Grela et al. regarding the catalytic activity of achiral 2a54 and 2b
(Fig. 8).55 The validity of such proposals in relation to chiral Ru complexes would be further substantiated if 3b proved to be significantly less active than 3. Complex 3d was investigated to establish whether a recent observation regarding higher activity of its corresponding achiral analogue 2c
(Fig. 8) pertains to the present class of chiral Ru catalysts.56 Enantiomerically pure Ru carbenes 3e and 3f were prepared to determine the influence of reduced electron donation to the Ru center by the aryloxide oxygen on catalytic activity.57
 |
| | Fig. 8 Modified versions of Ru complex 2. | |
 |
| | Scheme 20 Steric and electronic modifications of chiral Ru complex 3 leads to significantly more reactive catalysts 3d and 3f. | |
Study of catalytic activity of the modified chiral Ru complexes shown in Scheme 20 led us to establish that catalysts 3d and 3f exhibit reactivity levels that are more than two orders of magnitude higher than3. The relative rate data shown in Scheme 20 are illustrative.
The availability of the more effective chiral complexes has led to new possibilities in catalytic asymmetric olefin metathesis. Two examples are shown in Scheme 21. Whereas Ru-catalyzed AROM/CM of 47 leads to <10% conversion with 3
(and likely results in rapid polymerization with chiral Mo catalysts),58 in the presence of 10 mol%
3d, diamide 48a is generated in 92% ee and 65% isolated yield. Reaction of triene 49 cannot be promoted in the presence of chiral Ru complex 3. However, in the presence of 10 mol%
3d, asymmetric RCM proceeds to >98% conversion to afford 50 in 76% ee. It should be noted that, thus far, chiral Mo-based catalysts are able to promote enantioselective RCM more effectively than either available class of chiral Ru catalysts;46,59 on the other hand, reactions with 3 or 3d can be carried out in air and with undistilled solvents.
 |
| | Scheme 21 Representative Ru-catalyzed enantioselective olefin metathesis reactions made possible by the more reactive complex 3d. | |
6 Conclusion and perspectives
The discovery of Ru complex 1 in 1996 and subsequent synthesis and investigation of the catalytic activity of non-phosphine catalyst 2 has yielded a class of user-friendly and practical olefin metathesis catalysts that offer unique levels of reactivity and selectivity. Of particular significance is the absence of a phosphine ligand in complex 2, an attribute that is largely responsible for its signature reactivity profile. Another important chracteristic, shared by Ru catalysts represented by 1–3, is the presence of an aryl ether ligating group which, not only provides a convenient handle for attachment of the transition metal complex to various solid supports but also allows for facile steric and electronic modifications of the catalyst structure. Increasingly effective achiral and chiral catalysts and their supported versions thus continue to be developed that are based on the structural platforms provided by complexes 1 and 2. Representative examples provided in this article indicate that the community of synthetic chemists is becoming increasingly aware of the special activity of this class of Ru catalysts. Furthermore, indications are beginning to appear in the literature that the field of polymer chemistry may soon follow suit.60
A number of critical issues remain to be addressed, despite the advances made in the past several years. Design, synthesis and development of more effective chiral Ru-based catalysts that promote a wider range of ring-closing, ring-opening or cross metathesis reactions should probably be placed at the top of this priority list. Although significant strides have been taken in enhancing the catalytic activity of Ru-based complexes, 1–10 mol% loadings are still required for efficient catalysis. Such conditions may be acceptable in small scale laboratory experiments but present a notable economic challenge in large scale synthesis. Judging from the remarkable developments of the recent past, it is likely that many more exciting and important advances will be forthcoming in this important area.
Acknowledgements
Financial support was provided by the NSF (CHE-9632278, CHE-9905806 and CHE-0213009) and Boehringer-Ingelheim. J. S. K. was supported as an NSF predoctoral fellow. We are grateful to Brian L. Gray, John D. Gleason and Jonathan M. Giftos for experimental assistance.
References
- For reviews on catalytic olefin metathesis, see:
(a) R. H. Grubbs, S. J. Miller and G. C. Fu, Acc. Chem. Res., 1995, 28, 446–452 CrossRef CAS;
(b) H-G. Schmalz, Angew. Chem., Int. Ed. Engl., 1995, 34, 1833–1836 CrossRef CAS;
(c) M. Schuster and S. Blechert, Angew. Chem., Int. Ed. Engl., 1997, 36, 2036–2056 CrossRef;
(d)
Alkene Metathesis in Organic Synthesis, ed. A. Furstner,Springer, Berlin, 1998 Search PubMed;
(e) S. K. Armstrong, J. Chem. Soc., Perkin Trans. 1, 1998, 371–388 RSC;
(f) R. H. Grubbs and S. Chang, Tetrahedron, 1998, 54, 4413–4450 CrossRef CAS;
(g) A. J. Phillips and A. D. Abell, Aldrichimica Acta, 1999, 32, 75–90 Search PubMed;
(h) A. Furstner, Angew. Chem., Int. Ed., 2000, 39, 3012–3043 CrossRef CAS;
(i) T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS;
(j) R. R. Schrock and A. H. Hoveyda, Angew. Chem., Int. Ed., 2003, 42, 4592–4633 CrossRef CAS.
- J. P. A. Harrity, M. S. Visser, J. D. Gleason and A. H. Hoveyda, J. Am. Chem. Soc., 1997, 119, 1488–1489 CrossRef CAS.
- J. P. A. Harrity, D. S. La, D. R. Cefalo, M. S. Visser and A. H. Hoveyda, J. Am. Chem. Soc., 1998, 120, 2343–2351 CrossRef CAS.
-
(a) P. Schwab, M. B. France, J. W. Ziller and R. H. Grubbs, Angew. Chem., Int. Ed. Engl., 1995, 34, 2039–2041 CrossRef CAS;
(b) P. Schwab, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1996, 118, 100–110 CrossRef CAS.
-
(a) R. R. Schrock, J. S. Murdzek, G. C. Bazan, J. Robbins, M. DiMare and M. O'Regan, J. Am. Chem. Soc., 1990, 112, 3875–3886 CrossRef CAS;
(b) G. C. Bazan, R. R. Schrock, H-N. Cho and V. C. Gibson, Macromolecules, 1991, 24, 4495–4502 CrossRef CAS.
- C. W. Johannes, M. S. Visser, G. S. Weatherhead and A. H. Hoveyda, J. Am. Chem. Soc., 1998, 120, 8340–8347 CrossRef CAS.
- The first example of a catalytically active mono-phosphine Ru complex was reported by Snapper and coworkers: J. A. Tallarico, P. J. Bonitatebus and M. L. Snapper, J. Am. Chem. Soc., 1997, 119, 7157–7158 Search PubMed.
- J. S. Kingsbury, J. P. A. Harrity and A. H. Hoveyda, J. Am. Chem. Soc., 1999, 121, 791–799 CrossRef CAS.
- E. L. Dias and R. H. Grubbs, Organometallics, 1998, 17, 2758–2767 CrossRef CAS.
- For application of the release/return mechanism to the design of a supported Ru catalyst. see: M. Ahmed, A. G. M. Barrett, D. C. Braddock, S. M. Cramp and P. A. Procopiou, Tetrahedron Lett., 1999, 40, 8657–8662 Search PubMed.
- E. L. Dias, S. T. Nguyen and R. H. Grubbs, J. Am. Chem. Soc., 1997, 119, 3887–3897 CrossRef CAS.
- S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168–8179 CrossRef CAS.
-
(a) M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956 CrossRef CAS;
(b) A. K. Chatterjee and R. H. Grubbs, Org. Lett., 1999, 1, 1751–1753 CrossRef CAS.
-
(a) T. Weskamp, W. C. Schattenmann, M. Spiegler and W. A. Hermann, Angew. Chem., Int. Ed., 1998, 37, 2490–2493 CrossRef CAS;
(b) T. Weskamp, F. J. Kohl, W. Hieringer, D. Gleich and W. A. Hermmann, Angew. Chem., Int. Ed., 1999, 38, 2416–2419 CrossRef CAS;
(c) M. Scholl, T. M. Trnka, J. P. Morgan and R. H. Grubbs, Tetrahedron Lett., 1999, 40, 2247–2250 CrossRef CAS;
(d) J. Huang, E. D. Stevens, S. P. Nolan and J. L. Petersen, J. Am. Chem. Soc., 1999, 121, 2647–2678 CrossRef;
(e) L. Ackermann, A. Furstner, T. Weskamp, F. J. Kohl and W. A. Hermann, Tetrahedron Lett., 1999, 40, 4787–4790 CrossRef CAS;
(f) A. Furstner, O. R. Thiel, L. Ackermann, H-J. Schanz and S. P. Nolan, J. Org. Chem., 2000, 65, 2204–2207 CrossRef.
- For reviews of nucleophilic carbenes, see:
(a) M. Regitz, Angew. Chem., Int. Ed., 1996, 35, 725–728 CrossRef CAS;
(b) W. A. Herrmann and C. Kocher, Angew. Chem., Int. Ed., 1997, 36, 2162–2187 CrossRef CAS;
(c) A. J. Arduengo and R. Krafczyk, Chem. Z., 1998, 32, 6–14 Search PubMed;
(d) A. J. Arduengo, Acc. Chem. Res., 1999, 32, 913–921 CrossRef.
- For a report on the accelerating effect of an α-hydroxyl group on Ru-catalyzed RCM reactions, see: T. R. Hoye and H. Zhao, Org. Lett., 1999, 1, 1123–1125 Search PubMed.
- For examples where 2 and 17 are reported to exhibit similar levels of efficiency (involving cross metathesis reactions), see:
(a) A. J. Geissert, L. Snyder, J. Markham and S. T. Diver, Org. Lett., 2003, 5, 1793–1796 CrossRef;
(b) O. M. Demchuk, K. M. Pietrusiewicz, A. Michrowska and K. Grela, Org. Lett., 2003, 5, 3217–3220 CrossRef CAS.
-
(a) S. BouzBouz and J. Cossy, Org. Lett., 2001, 3, 1451–1454 CrossRef CAS;
(b) J. Cossy, S. BouzBouz and A. H. Hoveyda, J. Organomet. Chem., 2001, 624, 327–332 CrossRef CAS.
- M. S. Sanford, J. A. Love and R. H. Grubbs, J. Am.Chem. Soc., 2001, 123, 6543–6554 CrossRef CAS.
-
(a) S. Imhof, S Randl and S. Blechert, Chem. Commun., 2001, 1692–1693 RSC;
(b) S. Randl, S. Gessler, H. Wakamatsu and S. Blechert, Synlett, 2001, 430–432 CAS;
(c) S. Randl, S. J. Connon and S. Blechert, Chem. Commun., 2001, 1796–1797 RSC;
(d) S. Gessler, S. Randl and S. Blechert, Tetrahedron Lett., 2000, 41, 9973–9976 CrossRef CAS.
- H. Mizutani, S. J. Degrado and A. H. Hoveyda, J. Am. Chem. Soc., 2001, 123, 779–780 CrossRef CAS.
- S. BouzBouz, E. De Lemos and J. Cossy, Adv. Synth. Catal., 2002, 344, 627–630 CrossRef CAS.
- F. Royer, C. Vilain, L. Ekraim and L. Grimaud, Org. Lett., 2003, 5, 2007–2009 CrossRef CAS.
-
(a) T. Naota, H. Takaya and S-I. Murahashi, Chem. Rev., 1998, 98, 2599–2660 CrossRef CAS;
(b) B. M. Trost, F. D. Toste and A. B. Pinkerton, Chem. Rev., 2001, 101, 2067–2096 CrossRef CAS.
- For other Ru-catalyzed tandem protocols, where one of the transformations is an olefin metathesis, see:
(a) C. W. Bielawski, J. Louie and R. H. Grubbs, J. Am. Chem. Soc., 2000, 122, 12872–12873 CrossRef CAS;
(b) A. E. Sutton, B. A. Seigal, D. F. Finnegan and M. L. Snapper, J. Am. Chem. Soc., 2002, 124, 13390–13391 CrossRef CAS;
(c) C. Cadot, P. Dalko and J. Cossy, Tetrahedron Lett., 2002, 43, 1839–1841 CrossRef CAS;
(d) M. Arisawa, Y. Terada, M. Nakagawa and A. Nishida, Angew. Chem., Int. Ed., 2002, 41, 4732–4734 CrossRef CAS.
-
(a) J. Cossy, F. C. Bargiggia and S. BouzBouz, Tetrahedron Lett., 2002, 43, 6715–6717 CrossRef CAS;
(b) J. Cossy, F. Bargiggia and S. BouzBouz, Org. Lett., 2003, 5, 459–462 CrossRef CAS.
-
(a) S. BouzBouz and J. Cossy, Org. Lett., 2001, 3, 1451–1454 CrossRef CAS;
(b) J. Cossy, S. BouzBouz, F. Pradaux, C. Willis and V. Bellosta, Synlett, 2002, 1595–1606 CrossRef CAS.
- K. E. Murphy and A. H. Hoveyda, J. Am. Chem. Soc., 2003, 125, 4690–4691 CrossRef CAS.
- For recent key reports and reviews regarding catalytic CM reactions, see:
(a) H. E. Blackwell, D. J. O'Leary, A. K. Chatterjee, R. A. Washenfelder, D. A. Bussmann and R. H. Grubbs, J. Am. Chem. Soc., 2000, 122, 58–71 CrossRef CAS;
(b) A. K. Chatterjee, T-L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360–11370 CrossRef CAS; S. J. Cannon and S. Blechert, Angew. Chem., Int. Ed., 2003, 42, 1900–1923 CrossRef.
- T. Lazarova, J. S. Chen, B. Hamann, J. M. Kang, D. Homuth-Trombino, F. Han, E. Hoffmann, C. McClure, J. Eckstein and Y. S. Or, J. Med. Chem., 2003, 46, 674–676 CrossRef CAS.
- P. Wipf, S. R. Rector and H. Takahashi, J. Am. Chem. Soc., 2002, 124, 14848–14849 CrossRef CAS.
- R. R. Cesati, J. de Armas and A. H. Hoveyda, J. Am. Chem. Soc., 2003, 125, in press.
- K. J. Hale, M. M. Domostoj, D. A. Tocher, E. Irving and F. Scheinmann, Org. Lett., 2003, 5, 2927–2930 CrossRef CAS.
- B. Nosse, R. B. Chhor, W. B. Jeong, C. Bohm and O. Reiser, Org. Lett., 2003, 5, 941–944 CrossRef CAS.
- For a more recent non-phosphine Ru-based metathesis catalyst and discussion of related mechanistic issues, see: J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, Angew. Chem., Int. Ed., 2002, 41, 4035–4037 Search PubMed.
- T. M. Trnka, M. W. Day and R. H. Grubbs, Angew. Chem., Int. Ed., 2001, 40, 3441–3444 CrossRef CAS.
- For a comprehensive review of supported catalysts for olefin metathesis, see:
J. S. Kingsbury, A. H. Hoveyda, in Polymeric Materials in Organic Synthesis and Catalysis, ed. M. R. Buchmeiser, Wiley-VCH, Weinheim, 2003, pp. 467–502 Search PubMed.
- Q. Yao, Angew. Chem., Int. Ed., 2000, 39, 3896–3898 CrossRef CAS.
- J. Dowden and J. Savovic, Chem. Commun., 2001, 37–38 RSC.
- Q. Yao and Y. Zhang, Angew. Chem., Int. Ed., 2003, 42, 3395–3398 CrossRef CAS.
- Sol-Gel Science: The Physics and Chemistry of Sol-Gel Processing, C. J.
Brinkler and G. W. Scherer,Academic Press, San Diego, CA, 1990.
- J. S. Kingsbury, S. B. Garber, J. M. Giftos, B. L. Gray, M. M. Okamoto, R. A. Farrer, J. T. Fourkas and A. H. Hoveyda, Angew. Chem., Int. Ed., 2001, 40, 4251–4256 CrossRef CAS.
- S. Randl, N. Buschmann, S. J. Connon and S. Blechert, Synlett, 2001, 1547–1550 CrossRef CAS.
- K. Grela, M. Tryznowski and M. Bieniek, Tetrahedron Lett., 2002, 43, 9055–9059 CrossRef CAS.
- S. J. Connon, A. M. Dunne and S. Blechert, Angew. Chem., Int. Ed., 2002, 41, 3835–3838 CrossRef CAS.
- For reviews of Mo-catalyzed enantioselective olefin metathesis, see:
(a) A. H. Hoveyda and R. R. Schrock, Chem. Eur. J., 2001, 7, 945–950 CrossRef CAS;
(b) R. R. Schrock and A. H. Hoveyda, Angew. Chem., Int. Ed., 2003, 42, 4592–4633 CrossRef CAS.
- J. J. VanVeldhuizen, S. B. Garber, J. S. Kingsbury and A. H. Hoveyda, J. Am. Chem. Soc., 2002, 124, 4954–4955 CrossRef CAS.
- For an alternative chiral Ru-based metathesis catalyst, see: T. J. Seiders, D. W. Ward and R. H. Grubbs, Org. Lett., 2001, 3, 3225–3228 Search PubMed.
- For a review of complexes chiral at the metal, see: H. Brunner, Angew. Chem., Int. Ed., 1999, 38, 1194–1208 Search PubMed.
- For an example regarding the utility of achiral non-phosphine complex 2 in ROM/CM, see: B. H. White and M. L. Snapper, J. Am. Chem. Soc., 2003, 125, in press Search PubMed.
-
(a) D. S. La, J. G. Ford, E. S. Sattely, J. P. Bonitatebus, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 1999, 121, 11603–11604 CrossRef CAS;
(b) D. S. La, E. S. Sattely, J. G. Ford, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2001, 123, 7767–7778 CrossRef CAS.
- For a related case, see: S. Chang, L. Jones, C. Wang, L. M. Henling and R. H. Grubbs, Organometallics, 1998, 17, 3460–3465 Search PubMed.
- J. J. Van Veldhuizen, D. G. Gillingham, S. B. Garber, O. Kataoka and A. H. Hoveyda, J. Am. Chem. Soc., 2003, 125, 12502–12508 CrossRef CAS.
- K. Grela, S. Harutyunyan and A. Michrowska, Angew. Chem., Int. Ed., 2002, 41, 4038–4040 CrossRef CAS.
- K. Grela and M. Kim, Eur. J. Org. Chem., 2003, 963–966 CrossRef CAS.
- H. Wakamatsu and S. Blechert, Angew. Chem., Int. Ed., 2002, 41, 2403–2405 CrossRef CAS.
- M. S. Sanford, L. M. Henling, M. W. Day and R. H. Grubbs, Angew. Chem., Int. Ed., 2000, 39, 3451–3453 CrossRef CAS.
- For representative reports on Mo-catalyzed AROM/CM, see:
(a) G. S. Weatherhead, J. G. Ford, E. J. Alexanian, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 1828–1829 CrossRef CAS;
(b) Ref. 51b;
(c) W. C. P. Tsang, J. A. Jernelius, G. A. Cortez, G. S. Weatherhead, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2003, 125, 2591–2596 CrossRef CAS.
- For examples of Mo-catalyzed ARCM, see:
(a) J. B. Alexander, D. S. La, D. R. Cefalo, A. H. Hoveyda and R. R. Schrock, J. Am. Chem. Soc., 1998, 120, 4041–4042 CrossRef CAS;
(b) D. S. La, J. B. Alexander, D. R. Cefalo, D. D. Graf, A. H. Hoveyda and R. R. Schrock, J. Am. Chem. Soc., 1998, 120, 9720–9721 CrossRef CAS;
(c) S. S. Zhu, D. R. Cefalo, D. S. La, J. Y. Jamieson, W. M. Davis, A. H. Hoveyda and R. R. Schrock, J. Am. Chem. Soc., 1999, 121, 8251–8259 CrossRef CAS;
(d) D. R. Cefalo, A. F. Kiely, M. Wuchrer, J. Y. Jamieson, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2001, 123, 3139–3140 CrossRef CAS;
(e) A. F. Keily, J. A. Jernelius, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2002, 124, 2868–2869 CrossRef CAS.
- For a recent report where Ru catalyst 2 is reported to be an “especially effective” initiator in polymer synthesis, see: S. Demel, S. Riegler, K. Wewerka, W. Schoefberger, C. Slugovc and F. Selzer, Inorg. Chim. Acta, 2003, 345, 363–366 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2004 |
Click here to see how this site uses Cookies. View our privacy policy here. 
