Understanding the puzzling chemistry of bicyclo[2.1.0]pentane†
Barry K.
Carpenter
Department of Chemistry and Chemical Biology, Baker Laboratory, Cornell University, Ithaca, NY 14853-1301, USA. E-mail: bkc1@cornell.edu; Fax: + 1 607 255 4137; Tel: + 1 607 255 3826
First published on 13th November 2003
Abstract
Three thermal reactions of bicyclo[2.1.0]pentane have been studied by CASPT2–g3 and CASSCF electronic structure calculations. They are isomerization to cyclopentene, isomerization to 1,4-pentadiene, and cycloaddition to fumaronitrile. All three of these reactions exhibit unusual features that have prompted mechanistic debate. The present computational results provide a basis for understanding the experimental observations.
Introduction
The report of the synthesis of bicyclo[2.1.0]pentane by Criegee and Rimmelin1 was followed in succeeding decades by a thorough study of its unimolecular and bimolecular thermal reactions. Several of these reactions have turned out to exhibit unusual features that have become the subject of mechanistic speculation and sometimes controversy. The calculations described in this article are designed to provide some insight into the origins of the observed phenomena.In 1962 Halberstadt and Chesick2 reported that bicyclo[2.1.0]pentane would isomerize to cyclopentene with an Arrhenius activation energy of 195.0 ± 4.2 kJ mol−1. Steel et al. later obtained a value of 190.7 ± 1.7 kJ mol−1 for the same quantity.3 Also in 1962 Chesick discovered that exo- and endo-2-methylbicyclo[2.1.0]pentane would interconvert with activation energies of 161.7 ± 3.3 kJ mol−1 (exo → endo) and 163.8 ± 3.3 kJ mol−1 (endo → exo).4 Subsequently, Baldwin and Ollerenshaw obtained a value of 158.2 ± 0.4 kJ mol−1 for the activation energy of interconversion of exo- and endo-bicyclo[2.1.0]pentane-cis-2,3-d2.5 Taken together, these data show that the isomerization of bicyclo[2.1.0]pentane to cyclopentene involves an activation barrier that is 34.7 ± 5.0 kJ mol−1 higher than that for exo–endo interconversion.
Probably the simplest mechanism for the stereochemical and structural isomerizations involves the scission of the C1–C4 bond of bicyclo[2.1.0]pentane as the first step, generating cyclopentane-1,3-diyl, presumably in a singlet electronic state. Reclosure or [1,2] hydrogen migration from this intermediate would then provide pathways for both observed reactions. Such a mechanism would be analogous to that proposed for the stereomutation and structural isomerization of cyclopropane, for which the singlet state of the trimethylene biradical is believed to be a common intermediate.6 However, the formation of propene from cyclopropane has an activation energy that differs from that for stereomutation by just 4–15 kJ mol−1.7 If the mechanisms are right, it is not clear why the hydrogen migration would be more difficult in cyclopentane-1,3-diyl than in trimethylene. (In principle, the results could also be explained if the reclosure of trimethylene faced a barrier significantly higher than that for reclosure of cyclopentane-1,3-diyl, but such an explanation would be inconsistent with experiments and calculations suggesting that the reclosure of singlet trimethylene is essentially barrierless, vide infra.)
One resolution of the apparent discrepancy between the reactions of cyclopropane and bicyclo[2.1.0]pentane would be to propose that, despite their apparent similarity, the formation of propene from cyclopropane and cyclopentene from bicyclo[2.1.0]pentane actually occur by different mechanisms. Baldwin and Andrews did consider such a possibility, recognizing that bicyclo[2.1.0]pentane could be converted to cyclopentene by a formal σ2s + σ2a cycloaddition between the C1–C2 and C4–C5 bonds.8 However, their experiments with bicyclo[2.1.0]pentane-5,5-d2 revealed unambiguously that this mechanism is not correct, and that the isomerization to cyclopentene does involve a 1,2-shift of a hydrogen from C5 of the starting material (see Fig. 1). Hence, to date, the puzzling difference between the cyclopropane and bicyclo[2.1.0]pentane isomerizations remains unexplained.
![Two mechanisms for the isomerization of bicyclo[2.1.0]pentane to cyclopentene, and their distinction through deuterium labeling.](/image/article/2004/OB/b310676d/b310676d-f1.gif) | ||
| Fig. 1 Two mechanisms for the isomerization of bicyclo[2.1.0]pentane to cyclopentene, and their distinction through deuterium labeling. | ||
At higher temperatures, bicyclo[2.1.0]pentane undergoes isomerization to 1,4-pentadiene. Steel et al.3 found an activation energy of 218.8 ± 2.5 kJ mol−1 for the rearrangement. This reaction also has some puzzling features. The crucial experimental observation came from Berson et al.9 who studied the stereochemistry of the reaction for the various diastereomers of 2,3-dimethylbicyclo[2.1.0]pentane. They concluded that the reaction took place with about a 10 : 1 preference for the stereochemistry that would be classified as σ2s + σ2a if the process were a concerted retro-2 + 2 reaction, or conrotatory if the reaction occurred from singlet cyclopentane-1,3-diyl as the first-formed intermediate. However, neither of these mechanistic explanations is entirely satisfactory. While it is true that σ2s + σ2a is the thermally allowed stereochemistry for the concerted process, it would be very surprising if the conversion of bicyclo[2.1.0]pentane to 1,4-pentadiene followed a concerted path when all of the experimental and computational evidence suggests that there is no concerted component to the analogous reaction of cyclobutane.10–13 For cyclobutane, the two C–C bonds to be broken are related by symmetry, whereas for bicyclo[2.1.0]pentane, the C1–C4 and C2–C3 bonds must have significantly different dissociation energies because of the differences in ring-strain released when they break. Surely this asymmetry ought to bias the bicyclic molecule more in the direction of asynchronous bond cleavage. On the other hand, a mechanism occurring, once again, via a cyclopentane-1,3-diyl intermediate has its own problems. For that mechanism it is difficult to explain why there would be any significant stereoselectivity at all in the cleavage of the C2–C3 bond, and even more difficult to explain why any preference that there may be should be for the conrotatory mode.
Analysis of the stereochemistry of C2–C3 cleavage in cyclopentane-1,3-diyl by use of an orbital correlation diagram reveals that the preference should depend on the ordering of and energy difference between the two symmetry-adapted linear combinations of the singly occupied p-type orbitals on C1 and C4. (Unfortunately, the IUPAC rules of nomenclature change the numbering of the carbons after the C1–C4 bond of bicyclo[2.1.0]pentane is broken. In an effort to minimize confusion, the bicyclopentane numbering system is used throughout this discussion.) Ab initio electronic structure calculations have revealed that the biradical has a C2-symmetry minimum-energy geometry.14 In this point group, the nominally singly occupied p-type orbitals form a and b-symmetry combinations. If the a orbital were significantly lower in energy than the b orbital, then the preferred mode of C2–C3 cleavage would be disrotatory (see Fig. 2). If b were far below a, then a conrotatory cleavage would be preferred. In trimethylene – the biradical created by homolysis of one C–C bond of cyclopropane – calculations suggest that interaction of the p-type orbitals with the C–H orbitals of the central methylene causes the a combination to be lower in energy.15 The same effect should be in evidence for cyclopentane-1,3-diyl. However, the distance between the radical sites is a little smaller for cyclopentane-1,3-diyl than for trimethylene. The smaller distance strengthens the through-space interaction of the p-type basis orbitals, which lowers the energy of the b combination.15,16 According to the calculations of Conrad et al.17 these contributing factors almost perfectly cancel, so that in a two-configuration wavefunction – the minimum necessary to describe a singlet biradical – the a2 and b2 configurations have almost identical weights. If that prediction were correct, there should be no orbital-symmetry-derived preference for disrotatory or conrotatory cleavage of the C2–C3 bond.
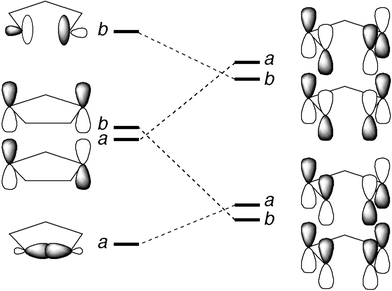 | ||
| Fig. 2 Orbital correlation diagram for the conrotatory ring opening of cyclopentane-1,3-diyl. Four electrons need to be correlated by this diagram. If the a combination of p-type orbitals in cyclopentane-1,3-diyl is far below the b combination, then the conrotatory ring opening should be forbidden. If the a and b combinations are very close in energy (as ab initio calculations suggest) then the correlation diagram would predict that there should be little or no stereoselectivity to the reaction. | ||
Perhaps the most mysterious-seeming of all the reactions of bicyclo[2.1.0]pentane is its cycloaddition to electron-deficient alkenes. In 1968, Gassman et al.18 reported that such a reaction would occur when the hydrocarbon was heated with maleonitrile or fumaronitrile. Among the products were dicyanonorbornanes, arising from a formal 2 + 2 cycloaddition across the C1–C4 bond. When bicyclo[2.1.0]pentane-exo,exo-2,3-d2 was used in the reaction the really surprising aspect of this reaction was revealed: the cycloadducts were found also to have the labels in the exo sites, indicating that the cycloaddition had occurred exclusively from the endo face of the hydrocarbon reactant. Not only is this the more sterically hindered face, it is also the one that would seem to give the poorer access to the electron density of the C1–C4 bond. Since the cycloaddition apparently occurs only with electron-deficient alkenes, it seems reasonable to assume that the alkene is acting as the more electrophilic reagent, and one might have thought that it would consequently seek the site of highest electron density in the bond with which it is reacting. Calculations at all levels confirm that this is definitely on the exo face of bicyclo[2.1.0]pentane, just as a conventional “banana-bond” model would have led one to expect. Nevertheless, the preference for endo addition is apparently strong, as revealed by experiments from this laboratory in which the addition partner (in this case an electron-deficient alkyne) was tethered to C5 of the bicyclo[2.1.0]pentane. These studies revealed that even when the alkyne was attached by just a –(CH2)3– chain to the exo site on C5, its addition to the C1–C4 bond occurred from the endo face.19
The reaction that is most frequently used to prepare bicyclo[2.1.0]pentane – deazetization of 2,3-diazabicyclo[2.2.1] hept-2-ene – looks at first sight as if it might provide some clues to the cycloaddition puzzle, because deuterium labeling reveals that it occurs with a preference for inversion. Originally20 the ratio of inversion to retention for the thermal reaction was believed to be 3:1, but correction for epimerization of the bicyclopentane stereoisomers reveals it to be 4.7 ± 0.9:1.21 The overall stereochemistry of the nitrogen extrusion thus makes the process look like a “microscopic reverse” of the cycloaddition reactions.22 However, a detailed experimental and computational study of the deazetization has provided evidence that the reaction occurs by synchronous C–N bond scission to give a labeled cyclopentane-1,3-diyl whose stereochemistry of closure is dictated by nonstatistical dynamical effects.23 In contrast, cyclopentane-1,3-diyl can be rigorously excluded as an intermediate in several of the cycloadditions, whose activation enthalpies are substantially lower than that for formation of the biradical from bicyclo[2.1.0]pentane.24 Hence one must conclude either that the proposed mechanism for the nitrogen extrusion is wrong, or that the similarity in over all stereochemistry between that reaction and the cycloadditions is coincidental. Further discussion on this point will be presented later in the article.
Results and discussion
Because singlet biradicals were anticipated to play prominent roles in most or all of the mechanisms to be studied, it seemed clear from the outset that a multiconfigurational computational model would be required for reliable descriptions of the intermediates and transition states. Among such methods, multireference second-order perturbation theory seems to strike a good balance between reliability and cost. For the present work, the CASPT2 method,25 with the so-called g3 correction to the zeroth-order Hamiltonian, was selected, since this model seems to provide a balanced treatment of electron correlation in closed and open-shell species.26 For most of the calculations, the Dunning cc-pVDZ basis set was used. However, for the addition of fumaronitrile to bicyclo[2.1.0]pentane, two considerations suggested a different basis set. The first was the need to consider a charge-transfer or even full electron-transfer step in the reaction, since a mechanism of this kind has been suggested as an explanation for the observed stereoselectivity.19 Were there to be even partial electron transfer from the bicyclo[2.1.0]pentane to the fumaronitrile, the latter would acquire anionic character, which would in turn suggest the need to employ a basis set that included diffuse functions in the calculations. However, the size of the system – the second consideration – made the use of aug-cc-pVDZ impractical, and so for this reaction the 6-31+G(d,p) basis set was selected.The geometries of the stationary points in the reactions of interest were optimized at the CASSCF(8,8) level. Single-point CASPT2–g3(8,8) calculations were then carried out on the optimized structures. For the unimolecular reactions of bicyclo[2.1.0]pentane, the active space consisted of the σ and σ* orbitals of the C1–C4 and C2–C3 bonds, and of the two C–H bonds on C5. Although not all of these orbitals would be actively involved in every reaction, it seemed likely that a comparison of activation barriers for the competitive reactions would be most reliable if all structures were calculated with the same active space.
For the fumaronitrile addition, the active space consisted of the σ and σ* orbitals of the C1–C4 bond of bicyclo[2.1.0]pentane, plus the π and π* orbitals of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of fumaronitrile and of the CN groups in conjugation with it. In order to ensure that the orthogonal π and π* orbitals on the nitriles could safely be omitted from the active space, single point CASSCF(12,12)/6-31+G(d,p) calculations were run on the reactants and on the addition transition state. The difference in potential energy with the larger active space differed from that found with the (8,8) active space by only 1.9 kJ mol−1, indicating that the smaller active space was probably adequate for the task.
C bond of fumaronitrile and of the CN groups in conjugation with it. In order to ensure that the orthogonal π and π* orbitals on the nitriles could safely be omitted from the active space, single point CASSCF(12,12)/6-31+G(d,p) calculations were run on the reactants and on the addition transition state. The difference in potential energy with the larger active space differed from that found with the (8,8) active space by only 1.9 kJ mol−1, indicating that the smaller active space was probably adequate for the task.
Isomerization to cyclopentene
Since the labeling studies of Baldwin and Andrews had essentially defined the mechanism of this reaction,8 the question to be addressed was whether the calculations could reproduce the observed difference in activation barrier for reclosure and hydrogen migration in cyclopentane-1,3-diyl, and, if so, whether they could afford insight into the difference between the bicyclo[2.1.0]pentane → cyclopentene and cyclopropane → propene isomerizations.The CASSCF geometry optimization of cyclopentane-1,3-diyl produced a C2-symmetry minimum, as previously reported.14,23 Its ring closure was found to occur via a C1-symmetry transition structure, and had an activation enthalpy of 5.3 kJ mol−1 from the CASPT2-g3 calculations. This value is very much in line with earlier results and with experimental estimates.23 The 1,2-hydrogen migration was found to have an activation enthalpy of 38.6 kJ mol−1. The experimental results revealed a difference in Arrhenius activation energies for these two reactions of 34.7 ± 5.0 kJ mol−1. If converted to an activation enthalpy difference, the value is almost the same: 34.6 ± 5.0 kJ mol−1, which is in excellent agreement with the calculated difference of 33.3 kJ mol−1.
For the conversion of trimethylene to propene, CASPT2–g3//CASSCF(6,6)/cc-pVDZ calculations found an activation enthalpy of 9.7 kJ mol−1. The conrotatory ring closure to cyclopropane was found to have a small potential energy barrier, but zero-point energy corrections made the ring closure barrierless, as has been found in other calculations.27 Consequently, the difference in barrier to stereomutation and structural isomerization of cyclopropane should be 9.7 kJ mol−1. Although this result is in excellent agreement with the experimental estimate, it should be noted that it differs significantly from the highest-level ab initio result reported previously. This is the MRCI calculation of Doubleday,28 who found that the hydrogen migration had a barrier 24.7 kJ mol−1 higher than that for stereomutation. In order to ensure that the present CASPT2 result was not spurious, a new, larger scale MRCI calculation was undertaken. Using the CASSCF(6,6)/cc-pVDZ geometries and ZPE corrections, a CISD + Q calculation was carried out from a CASSCF(6,6)/cc-pVTZ reference wavefunction. This calculation gave a difference in barrier heights of 16.4 kJ mol−1, suggesting that the CASPT2 result may be an underestimate, but not by as large a margin as the Doubleday calculation would have suggested.
The first conclusion from the calculations is that one can reproduce the difference in thermochemistry for the bicyclo[2.1.0]pentane → cyclopentene and cyclopropane → propene isomerizations without need to invoke different mechanisms. That is not a very interesting inference, since the labeling experiments of Baldwin and Andrews8 had more or less mandated the same conclusion, but it is a necessary outcome if the calculations are to be considered reliable. A more valuable step in the analysis is to find out why these similar mechanisms have different barriers. A possible clue can be found by comparing the CASSCF geometries for the transition structures (Fig. 3). In the parameters that one might think of as defining the reaction coordinate – the bond length and bond angle to the migrating hydrogen, and the lengths of the bonds from the radical carbons to their shared methylene – the structures are quite similar. However, in one coordinate that might initially have seemed unimportant – the CCC bond angle at the shared methylene – they are very different. In the trimethylene transition structure this angle is 128.0°, whereas in the cyclopentane-1,3-diyl transition structure it is 111.0°. The large angle in the trimethylene transition structure is particularly striking because its value exceeds those (114.6° and 125.3°, respectively) calculated for the trimethylene local minimum and for propene at the same level of theory. Its occurrence suggests that some repulsion may develop between the ends of the trimethylene chain during the hydrogen migration, and indeed a simple extension to Hoffmann's original orbital analysis of trimethylene suggests that that should be the case. His extended Hückel analysis of the frontier orbitals of trimethylene placed the a combination (using the C2 irreducible representations) of p-type orbitals on the termini below the b combination, because the latter was destabilized by an antibonding interaction with the C–H σ orbitals of the central methylene (Fig. 4).15 As described in the introduction, the a combination would be expected to rise in energy as the CCC angle decreases, whereas the b combination should drop. Were trimethylene well described by a single electronic configuration, this effect might be expected to lead to an unusually large CCC angle. However, in reality there are significant contributions from the a2 and b2 configurations. In the CASSCF calculations used here, their relative weights in the total wavefunction are 1.2 : 1. Presumably this effect tempers the tendency for CCC angle change because the two configurations respond in opposite ways.
![CASSCF/cc-pVDZ geometries for the [1,2] hydrogen migration transitions states in cyclopentane-1,3-diyl and trimethylene. The calculations used (8,8) and (6,6) active spaces, respectively.](/image/article/2004/OB/b310676d/b310676d-f3.gif) | ||
| Fig. 3 CASSCF/cc-pVDZ geometries for the [1,2] hydrogen migration transitions states in cyclopentane-1,3-diyl and trimethylene. The calculations used (8,8) and (6,6) active spaces, respectively. | ||
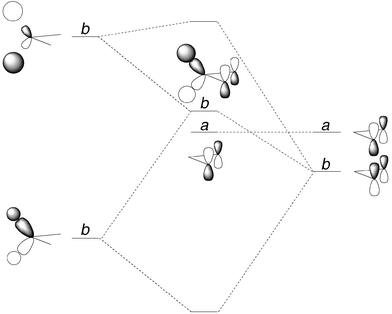 | ||
| Fig. 4 Interaction of the a- and b-symmetry combinations of trimethylene p-type orbitals with the σ and σ* orbitals of the central methylene. | ||
Once the hydrogen begins its migration, the picture changes significantly. Because the erstwhile b-symmetry (the point group is reduced to C1 when the hydrogen begins to migrate, but the C2 labels serve as useful identifiers) orbital contains an antibonding contribution from the hydrogen that is migrating – the analog of the antibonding interaction with the methylene C–H σ orbitals in trimethylene – the molecular orbital rises sharply in energy. However, the a-symmetry combination of p-type orbitals interacts weakly with the central methylene – in trimethylene it has zero interaction by symmetry – and so the net result is that the gap between the a and b orbitals increases (Fig. 5). This, in turn, causes the contribution from the a2 configuration to rise, becoming 5.5 : 1 with respect to b2 in the transition state, according to the present CASSCF calculations. It is presumably the higher occupancy of the a orbital in the H-migration transition structure that drives the expansion of the CCC angle.
![Increase in the gap between the frontier orbitals of trimethylene upon [1,2] migration of a hydrogen.](/image/article/2004/OB/b310676d/b310676d-f5.gif) | ||
| Fig. 5 Increase in the gap between the frontier orbitals of trimethylene upon [1,2] migration of a hydrogen. | ||
The extension of this analysis to cyclopentane-1,3-diyl is now obvious. The ring prevents significant angle expansion at the carbon from which the hydrogen is migrating (C5 in the original bicyclo[2.1.0]pentane numbering), and so the transition state for hydrogen migration is unable to benefit from an energy-lowering geometrical change that the analogous transition state for trimethylene can enjoy. This appears to be the principal source of the activation energy difference between these two similar rearrangements.
Isomerization to 1,4-pentadiene
For this reaction, the questions to be addressed by the calculations were whether the observed preference for the overall σ2s + σ2a stereochemistry could be reproduced, whether the reaction was calculated to occur with the correct activation enthalpy, and whether the mechanism was in fact a concerted retro 2 + 2 cycloaddition, or instead occurred with the intermediacy of cyclopentane-1,3-diyl. If the latter mechanism were found to be preferred, it would then be desirable to understand what controls the stereochemistry of C2–C3 cleavage.Two transition states for breaking the C2–C3 bond were located at the CASSCF level. One had C2 symmetry, corresponding to conrotation, and the other had Cs symmetry, corresponding to disrotation (Fig. 6). CASSCF zero-point-energy corrections, and CASPT2-g3 single-point dynamic correlation corrections provided a best estimate of ΔH° (Cs–C2) = 44.9 kJ mol−1. An intrinsic-reaction coordinate (IRC) calculation on the C2 transition structure showed that it connected cyclopentane-1,3-diyl to 1,4-pentadiene. It was found to be 54.3 kJ mol−1 higher in enthalpy than the transition state for ring closure of the biradical back to bicyclo[2.1.0]pentane – in reasonable accord with the experimental value of 60.6 ± 2.6 kJ mol−1.
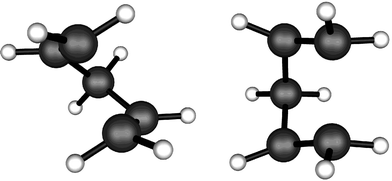 | ||
| Fig. 6 CASSCF(8,8)/cc-pVDZ transition structure for the conrotatory (C2) and disrotatory (CS) ring opening of cyclopentane-1,3-diyl to 1,4-pentadiene. | ||
The fact that the calculations find the right stereochemistry and right activation barrier for the reaction gives one some confidence that the mechanism is correct; that mechanism once again involves cyclopentane-1,3-diyl as an intermediate. Against expectations based on simple orbital-symmetry arguments, the calculations show a strong stereochemical preference in favor of conrotatory cleavage of the C2–C3 bond. In fact it is possible that the computed preference is too strong, since the 44.9 kJ mol−1 enthalpy difference between disrotatory and conrotatory pathways would lead to more than the 10 : 1 stereoselectivity observed by Berson et al.9 On the other hand, the experiments were conducted with 2,3-dimethylbicyclo[2.1.0]pentane, and so it is also possible that steric interactions of the methyl substituents with each other or with the ring could have reduced the intrinsic stereoselectivity.
As with the isomerization to cyclopentene, the most useful result of these calculations would be to provide some understanding of why the reaction occurs in the way that it does. A promising route to that goal comes from recognizing the similarity of the bicyclo[2.1.0]pentane and bicyclo[2.2.0]hexane ring-cleavage reactions.29 In both cases, the conversion to the diene product occurs with a preference for the formal σ2s + σ2a stereochemistry. Recent CASPT2 calculations by Hrovat and Borden30 have revealed that the ring cleavage of bicyclo[2.2.0]hexane begins with scission of the C1–C4 bond, to generate cyclohexane-1,4-diyl in a twist-boat conformation. In this conformation, the p-type orbitals at C1 and C4 are poorly aligned with the σ and σ* orbitals of the C2–C3 or C5–C6 bonds, and so ring cleavage to give 1,5-hexadiene would have a high barrier. However, the biradical is able to traverse a half-chair transition state on its way to a chair conformation. Interestingly, the orbital overlap in the chair is so good that there is no barrier to breaking either the C2–C3 or C5–C6 bond. In other words, the chair is itself a transition state – for the Cope rearrangement. It is a topological certainty that somewhere along a path connecting two transition states one must encounter a valley-ridge inflection point,31 and indeed Hrovat and Borden did find that an IRC calculation following the path down from the half chair, at some point before reaching the Cope transition state, breaks symmetry to give one of the two possible 1,5-hexadiene products. Nevertheless, it is accurate to say that it is the conformational change of the biradical from its initial twist-boat geometry to a near-chair geometry that is responsible for the observed formal σ2s + σ2a stereochemistry of the bicyclo[2.2.0]hexane → 1,5-hexadiene reaction.
However, there is an alternative conformation of cyclohexane-1,4-diyl that permits the orbital overlap required for ring opening; it is the boat conformation. Hrovat and Borden found the boat transition state for formation of 1,5-hexadiene. Again it is similar to, but not identical with the boat transition state for the Cope rearrangement. Were this the preferred pathway for the bicyclo[2.2.0]hexane cleavage, the reaction would have occurred with formal σ2s + σ2s stereochemistry. As we know from experiment, that is not the case, and in accord with that fact Hrovat and Borden found the boat transition state to be energetically disfavored, just as it is for the Cope rearrangement.
The geometrical similarity of the chair and boat pathways for bicyclo[2.2.0]hexane cleavage to those for the Cope rearrangement32 suggests that the explanations for their relative energies may also be similar. An explanation for the preferred stereochemistry of the Cope rearrangement was provided long ago by Hoffmann and Woodward.33 They pointed out that the two transition states could be hypothetically constructed by bringing together two allyl radicals in either a C2v (boat) or C2h (chair) geometry. If one traces the standard three π orbitals of each allyl fragment as they interact in these geometries, one discovers that the orbitals on the central carbons suffer an antibonding interaction in the boat structure, but not in the chair (because they are too far apart). The orbitals in question correspond to those on C1 and C4 of cyclohexane-1,4-diyl.
The potential relevance of the Hoffmann and Woodward analysis to the question at hand – the reason for the preferred conrotatory ring opening of cyclopentane-1,3-diyl – can be illustrated with the aid of a gedanken experiment, illustrated in Fig. 7. In this exercise one imagines pushing together the C5 and C6 methylenes of the chair and boat Cope transition states until they merge into one at the midpoint. This fanciful transformation converts the C2h (chair) structure into a C2 structure that looks a great deal like the calculated conrotatory transition state for ring opening of cyclopentane-1,3-diyl. Similarly, the C2v (boat) Cope transition state is transformed into a Cs structure that is very similar to the disrotatory transition state for ring opening of cyclopentane-1,3-diyl. The important point about these unphysical transformations is that they leave the p-type “radical” orbitals largely unaffected. Hence the antibonding interaction that disfavors the boat Cope transition state should also disfavor the disrotatory transition state for ring opening of cyclopentane-1,3-diyl. Supporting this analogy is the fact that the experimental32 enthalpy difference between the boat and chair Cope transition states (47 ± 8 kJ mol−1) is similar to the value calculated here (44.9 kJ mol−1) for the enthalpy difference between disrotatory and conrotatory transistion states for ring opening of cyclopentane-1,3-diyl.
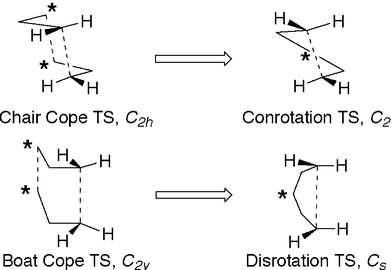 | ||
| Fig. 7 Conceptual relationship between the chair and boat transition states for the Cope rearrangement of 1,5-hexadiene and the conrotatory and disrotatory ring opening transition states of cyclopentane-1,3-diyl. In this gedanken experiment, the starred methylenes of the Cope transition states are pushed together symmetrically until they merge. | ||
It is somewhat amusing to recognize that at the end of all of this analysis, one has concluded that the principal factor disfavoring disrotatory ring opening of cyclopentane-1,3-diyl is exactly the same as the factor disfavoring the 1,2-hydrogen migration – the enforced antibonding interaction between the p-type “radical” orbitals in the transition states. In the case of the hydrogen migration there is no alternative, and so one just pays the price in a higher barrier. In the case of the ring opening there is an alternative – the system follows the conrotatory path in which this unfavorable interaction is minimized.
Cycloadditions
The questions of principal interest for cycloadditions to bicyclo[2.1.0]pentane are why reactions of this type occur exclusively from the endo face of the hydrocarbon in all of the cases that have been studied, and whether the stereoselectivity of nitrogen loss from 2,3-diazabicyclo[2.2.1]hept-2-ene-exo,exo-d2 (DBH-d2) can be explained by a mechanism that is a “microscopic reverse” of the cycloaddition.A thermochemical analysis helps to delineate some of the mechanistic parameters of these reactions. Experimental heats of formation34,35 reveal that the overall conversion of DBH to bicyclo[2.1.0]pentane + N2 is exothermic by 50 kJ mol−1 (Fig. 8). In striking contrast, the hypothetical conversion of norbornane to bicyclo[2.1.0]pentane + ethylene would be endothermic by 265 kJ mol−1 (Fig. 9). The addition of electron-withdrawing groups to the extruded alkene (in order to make the reaction a true microscopic reverse of an actual cycloaddition) has little effect. For example, the extrusion of maleic anhydride from the corresponding norbornane derivative is calculated to be endothermic by 273 kJ mol−1. The difference between the N2 and alkene extrusions is obviously the strong thermodynamic driving force for formation of molecular nitrogen. This, in turn, has an influence on the mechanism by which the fragmentations occur.
![Summary of the thermochemistry for deazetization of 2,3-diazabicyclo[2.2.1]hept-2-ene (DBH). The thermodynamic stability of N2 makes the overall reaction exothermic and favors synchronous scission of the C–N bonds, which produces N2 directly.](/image/article/2004/OB/b310676d/b310676d-f8.gif) | ||
| Fig. 8 Summary of the thermochemistry for deazetization of 2,3-diazabicyclo[2.2.1]hept-2-ene (DBH). The thermodynamic stability of N2 makes the overall reaction exothermic and favors synchronous scission of the C–N bonds, which produces N2 directly. | ||
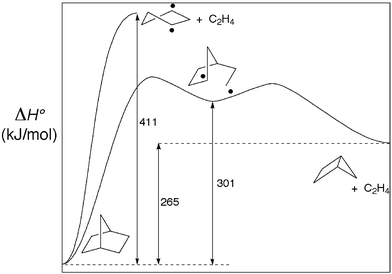 | ||
| Fig. 9 Summary of the thermochemistry for ethylene extrusion from norbornane. Unlike the formaally analogous deazetization of DBH (Fig. 8), this reaction is highly endothermic. Synchronous scission of the two C–C bonds is highly disfavored in this reaction. | ||
CASPT2 calculations on the thermal N2 extrusion from DBH suggest that the reaction occurs by synchronous scission of the two C–N bonds to generate singlet cyclopentane-1,3-diyl.23 Experimental heats of formation for DBH34 and for the triplet state of cyclopentane-1,3-diyl36 are known. Since all calculations that have been done suggest a singlet–triplet gap of about 4 kJ mol−1 for the biradical,14,17,37 it is probably safe to assume that the real value is somewhere close to that. Combining these pieces of information, one deduces that the conversion of DBH to cyclopentane-1,3-diyl + N2 is endothermic by 96 kJ mol−1. Since the experimental activation enthalpy for the reaction is 151.5 kJ mol−1,38 this pathway is kinetically viable. The CASPT2 calculations also showed that the alternative stepwise scission of the C–N bonds was less favorable by 30 kJ mol−1.23 Presumably this is because the synchronous process immediately releases the thermodynamically favorable N2 whereas the stepwise bond cleavage does not.
In contrast, experimental heats of formation show that the symmetrical conversion of norbornane to singlet cyclopentane-1,3-diyl + ethylene would be endothermic by 411 kJ mol−1. This value certainly exceeds any plausible estimate of the C1–C2 bond dissociation enthalpy in norbornane, since the C–C bond dissociation enthalpy in ethane is only 375 kJ mol−1. Consequently it is certain that norbornane would break one C–C bond rather than two. Again, the situation is not significantly changed if one considers maleic anhydride extrusion instead of ethylene extrusion. In this case, the overall endothermicity is estimated to be slightly higher at 419 kJ mol−1, but if anything the carbonyl groups would weaken the C–C bonds towards homolysis. The biradical generated by C1–C2 homolysis of norbornane could bypass cyclopentane-1,3-diyl by undergoing the intramolecular equivalent of an aliphatic radical substitution (SH2) reaction – affording ethylene and bicyclopentane directly. The stereochemistry of such reactions has been little studied, but in one related case (Cl atom addition to cyclopropane) it was found to occur with inversion.39 Were that to be the preferred mechanism in this case, the overall stereochemistry of the reaction would be in accord with that observed for cycloadditions to bicyclo[2.1.0]pentane.
The present CASPT2-g3 calculations support this picture. CASSCF(8,8)/6-31+G(d,p) calculations revealed the transition state for addition of fumaronitrile to bicyclo[2.1.0]pentane depicted in Fig. 10. An intrinsic reaction coordinate calculation showed it to be connected to fumaronitrile + bicyclo[2.1.0]pentane on one side and to a biradical local minimum on the other. The existence of such an intermediate is consistent with the experimental results showing loss of stereochemical integrity in the cycloadditions of fumaronitrile and maleonitrile to bicyclo[2.1.0]pentane.18 CASPT2-g3 single-point calculations, in addition to CASSCF ZPE and thermal corrections led to the prediction of an activation enthalpy of 127.5 kJ mol−1 for the addition. No experimental value for the activation enthalpy appears to have been determined, but it is noteworthy that the calculated barrier is smaller than that for epimerization of bicyclo[2.1.0]pentane, which is a result that is in accord with the experimental facts. One thus sees that the mysterious endo cycloaddition to bicyclo[2.1.0]pentane does not really occur from within the envelope flap of the hydrocarbon, but rather occurs by stepwise bond formation, beginning at one side. There is no evidence to support a previous proposal19 that significant electron transfer could occur between bicyclo[2.1.0]pentane and fumaronitrile during the reaction (at least in the gas phase) since the calculation of total Mulliken charges reveals a maximum of 0.15e transfer along the IRC.
![CASSCF(8,8)/6-31+G(d,p) transition structure for the addition of fumaronitrile to bicyclo[2.1.0]pentane. The reaction breaks the C1–C4 bond of the hydrocarbon with inversion and creates a biradical intermediate. When the second C–C bond is made, the overall addition appears to have occurred from the endo face of the bicyclo[2.1.0]pentane.](/image/article/2004/OB/b310676d/b310676d-f10.gif) | ||
| Fig. 10 CASSCF(8,8)/6-31+G(d,p) transition structure for the addition of fumaronitrile to bicyclo[2.1.0]pentane. The reaction breaks the C1–C4 bond of the hydrocarbon with inversion and creates a biradical intermediate. When the second C–C bond is made, the overall addition appears to have occurred from the endo face of the bicyclo[2.1.0]pentane. | ||
Conclusions
The present calculations have provided the following answers to the questions that were raised in the introduction.1. The barrier to 1,2-H migration for cyclopentane-1,3-diyl is higher than that for trimethylene because the five-membered ring enforces a destabilizing through-space interaction between the “radical” p-type orbitals in their a-symmetry linear combination. The a-symmetry combination becomes more heavily populated as the transition state for H migration is approached. In trimethylene, expansion of the CCC angle serves to reduce the through-space repulsion, but in cyclopentane-1,3-diyl this is impossible.
2. Exactly the same through-space interaction favors the conrotatory over the disrotatory stereochemistry for ring opening of cyclopentane-1,3-diyl. The phenomenon is essentially identical to the one favoring the chair over the boat transition state for the Cope rearrangement.
3. The apparent endo cycloaddition of fumaronitrile to bicyclo[2.1.0]pentane is found actually to occur by attack from the side, leading to the formation of a biradical intermediate. Despite the similarity in overall stereochemistry of the cycloaddition to that for the cycloreversion of 2,3-diazabicyclo-[2.2.1]hept-2-ene-d2, the mechanisms are found to be quite different. This difference is traceable largely to the thermodynamic stability of molecular nitrogen. In the cycloreversion direction the thermodynamics favor direct formation of N2 by synchronous bond scission, whereas the formally analogous extrusion of fumaronitrile from 2,3-dicyanonorbornane would be predicted to occur by stepwise bond cleavage. In the forward cycloaddition direction one can say that the thermodynamic stability of N2 gives its direct bimolecular reaction with bicyclo[2.1.0]pentane a barrier that is substantially higher than that for the unimolecular cleavage of the C1–C4 bond of the hydrocarbon. However, for fumaronitrile the cleavage of the C1–C4 bond by bimolecular addition is the lower-barrier pathway. This reaction occurs with the same stereochemistry (inversion) as that observed for Cl atom addition to cyclopropane.
Acknowledgements
Support of this research by the US National Science Foundation (Grant CHE-0212149) is gratefully acknowledged.References
- R. Criegee and A. Rimmelin, Chem. Ber., 1957, 90, 414 CAS.
- M. L. Halberstadt and J. P. Chesick, J. Am. Chem. Soc., 1962, 84, 2688 CrossRef CAS.
- C. Steel, R. Zand, P. Hurwitz and S. G. Cohen, J. Am. Chem. Soc., 1964, 86, 679 CrossRef CAS.
- J. P. Chesick, J. Am. Chem. Soc., 1962, 84, 3250 CrossRef CAS.
- J. E. Baldwin and J. Ollerenshaw, J. Org. Chem., 1981, 46, 2116 CrossRef CAS.
- E. W. Schlag and B. S. Rabinovitch, J. Am. Chem. Soc., 1960, 82, 5996 CrossRef CAS.
- E. V. Waage and B. S. Rabinovitch, J. Phys. Chem., 1972, 76, 1695 CrossRef CAS.
- J. E. Baldwin and G. D. Andrews, J. Org. Chem., 1973, 38, 1063 CrossRef CAS.
- J. A. Berson, W. Bauer and M. M. Campbell, J. Am. Chem. Soc., 1970, 92, 7515 CrossRef CAS.
- J. S. Chickos, J. Org. Chem., 1979, 44, 780 CrossRef CAS.
- J. S. Chickos, A. Annamalai and T. A. Keiderling, J. Am. Chem. Soc., 1986, 108, 4398 CrossRef CAS.
- M. J. Goldstein, M. J. Cannarsa, T. Kinoshita and R. F. Koniz, Stud. Org. Chem. (Amsterdam), 1987, 31, 121 Search PubMed.
- C. Doubleday Jr., J. Am. Chem. Soc., 1993, 115, 11968 CrossRef CAS.
- C. D. Sherrill, E. T. Seidl and H. F. Schaefer III, J. Phys. Chem., 1992, 96, 3712 CrossRef CAS.
- R. Hoffmann, J. Am. Chem. Soc., 1968, 90, 1475 CrossRef CAS.
- A. H. Goldberg and D. A. Dougherty, J. Am. Chem. Soc., 1983, 105, 284 CrossRef CAS.
- M. P. Conrad, R. M. Pitzer and H. F. Schaefer III, J. Am. Chem. Soc., 1979, 101, 2245 CrossRef CAS.
- P. G. Gassman, K. T. Mansfield and T. J. Murphy, J. Am. Chem. Soc., 1968, 90, 4746 CrossRef CAS.
- K. Redmond and B. K. Carpenter, J. Org. Chem., 1997, 62, 5668 CrossRef CAS.
- W. R. Roth and M. Martin, Justus Liebigs Ann. Chem., 1967, 702, 1 CAS.
- B. A. Lyons, J. Pfeifer, T. H. Peterson and B. K. Carpenter, J. Am. Chem. Soc., 1993, 115, 2427 CrossRef CAS.
- W. R. Roth and M. Martin, Tetrahedron Lett., 1967, 4695 CrossRef CAS.
- M. B. Reyes and B. K. Carpenter, J. Am. Chem. Soc., 2000, 122, 10163 CrossRef CAS.
- P. G. Gassman and K. T. Mansfield, J. Am. Chem. Soc., 1968, 90, 1517 CrossRef CAS.
- K. Andersson, M. Barysz, A. Bernhardsson, M. R. A. Blomberg, D. L. Cooper, M. P. Fülscher, C. de Graaf, B. A. Hess, G. Karlström, R. Lindh, P.-Å. Malmqvist, T. Nakajima, P. Neogrády, J. Olsen, B. O. Roos, B. Schimmelpfennig, M. Schütz, L. Seijo, L. Serrano-Andrés, P. E. M. Siegbahn, J. Stålring, T. Thorsteinsson, V. Veryazov and P.-O. Widmark, MOLCAS, Version 5.4, Lund University, Sweden, 2002 Search PubMed.
- K. Andersson, Theor. Chim. Acta, 1995, 91, 31 CrossRef CAS.
- S. J. Getty, E. R. Davidson and W. T. Borden, J. Am. Chem. Soc., 1992, 114, 2085 CrossRef CAS.
- C. Doubleday Jr., J. Phys. Chem., 1996, 100, 3520 CrossRef CAS.
- M. J. Goldstein and M. S. Benzon, J. Am. Chem. Soc., 1972, 94, 5119 CrossRef CAS.
- D. A. Hrovat and W. T. Borden, J. Am. Chem. Soc., 2001, 123, 4069 CrossRef CAS.
- P. Valtazanos and K. Ruedenberg, Theor. Chim. Acta, 1986, 69, 281 CrossRef CAS.
- M. J. Goldstein and M. S. Benzon, J. Am. Chem. Soc., 1972, 94, 7147 CrossRef CAS.
- R. Hoffmann and R. B. Woodward, J. Am. Chem. Soc., 1965, 87, 4389 CrossRef CAS.
- P. S. Engel, R. A. Melaugh, M. Mansson, J. W. Timberlake, A. W. Garner and F. D. Rossini, J. Chem. Thermodyn., 1976, 8, 607 CAS.
- R. B. Turner, P. Goebel, B. J. Mallon, W. v. E. Doering, J. F. Coburn Jr. and M. Pomerantz, J. Am. Chem. Soc., 1968, 90, 4315 CrossRef CAS.
- M. S. Herman and J. L. Goodman, J. Am. Chem. Soc., 1988, 110, 2681 CrossRef CAS.
- Y. Shao, M. Head-Gordon and A. I. Krylov, J. Chem. Phys., 2003, 118, 4807 CrossRef CAS.
- P. S. Engel, J. L. Wood, J. A. Sweet and J. L. Margrave, J. Am. Chem. Soc., 1974, 96, 2381 CrossRef CAS.
- J. H. Incremona and C. J. Upton, J. Am. Chem. Soc., 1972, 94, 301 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: geometries, harmonic vibration frequencies, energies, and natural orbital occupancies for all stationary points discussed in the paper. See http://www.rsc.org/suppdata/ob/b3/b310676d/ |
| This journal is © The Royal Society of Chemistry 2004 |