A ‘molecular switchboard’—covalent modifications to proteins and their impact on transcription†
Nelly Khidekel and Linda C. Hsieh-Wilson*
Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: lhw@caltech.edu
First published on 2nd December 2003
Abstract
Proteins undergo a remarkable variety of posttranslational modifications, with more than 200 distinct modifications identified to date. Increasing evidence suggests that many proteins bear multiple, distinct modifications, and the ability of one modification to antagonize or synergize the deposition of another can have significant biological consequences. Here, we illustrate the importance of posttranslational modifications within the context of transcriptional regulation, and we offer a perspective on the emerging role of combinatorial networks of modifications. Finally, we discuss the potential for chemical approaches to transform our understanding of the field.
 Nelly Khidekel Nelly Khidekel | Nelly Khidekel received her B.A. in chemistry from Northwestern University in 2000. She is currently pursuing her Ph.D. in the laboratory of Linda Hsieh-Wilson, where her work centers on understanding the role of covalent sugar modifications to proteins in the central nervous system. |
 Linda C. Hsieh-Wilson Linda C. Hsieh-Wilson | Linda Hsieh-Wilson received her B.S. in chemistry from Yale University in 1990. She was an NSF and ACS predoctoral fellow in the laboratory of Professor Peter G. Schultz at the University of California, Berkeley. In 1996, she moved to Rockefeller University to study neurobiology with Nobel Laureate Professor Paul Greengard as a Damon Runyon–Walter Winchell postdoctoral fellow. Linda joined the faculty at the California Institute of Technology in 2000, where her laboratory focuses on studying the impact of covalent modifications to proteins in the central nervous system. She has received a Beckman Young Investigator Award, an NSF CAREER Award, and an Alfred P. Sloan Fellowship. |
Introduction
A fundamental tenet of biochemistry is that proteins are composed of 20 basic building blocks. Given the limited range of chemical functionality present in the amino acid side chains, the diversity of protein structure and function is truly extraordinary. In reality, the situation is not so simple: Nature uses covalent modifications to proteins to complement and expand its chemical repertoire. Indeed more than 200 distinct posttranslational modifications (PTMs) have been identified to date, and the number and variety of modifications continue to grow (Fig. 1).1 Covalent modifications to proteins have been shown to have a profound impact at both the molecular and cellular level. For instance, the introduction of phosphoryl, glycosyl and other chemical groups can mediate the activity of a protein, affect its subcellular localization, or modulate its interaction with other macromolecules. At a more global level, PTMs have been shown to influence cellular events such as gene expression, cell cycle progression and programmed cell death. | ||
| Fig. 1 Some examples of posttranslational modifications. | ||
With an increased understanding of PTMs has come the exciting observation that many proteins undergo multiple, distinct chemical modifications. These discoveries point to the existence of complex combinatorial networks of PTMs with the potential to modulate different biological outcomes. Sorting out this ‘molecular switchboard’ of posttranslational events should reveal novel mechanisms for the control of biological systems and promises to open up new avenues for the exploration and manipulation of processes such as gene expression, signal transduction and cell-cycle progression. Here, we offer a perspective on this emerging area of research. Rather than provide an exhaustive survey, we have chosen to highlight the importance of PTMs in the specific context of transcription. In addition, we review examples of combinatorial networks of PTMs and discuss the potential for chemical approaches to transform our understanding of the field.
The process of transcription
The coordinated expression of specific genes is one of the most fundamental, exquisitely regulated processes in biology.2 For instance, the spatial and temporal expression of unique sets of genes differentiates a brain cell from a heart cell and enables each to perform its highly specialized functions. In the event of malfunction, aberrant gene expression can lead the cell down the path to cancer, neurodegenerative disorders and other diseases. Not surprisingly, organisms have evolved intricate mechanisms to achieve control and specificity over gene expression patterns. The molecular machinery responsible for gene transcription in eukaryotes consists of RNA polymerase II (RNAP II) and over 80 distinct regulatory proteins, which together initiate or suppress RNA synthesis at sites throughout the genome. Among the best understood factors are the sequence-specific transcription factors, which initiate gene-specific transcription and comprise an estimated 5–10% of the genes in the human genome. In addition, general transcription factors, along with coactivators and corepressors, assist the polymerase with catalysis and recognition of the DNA promoter sequence. Finally, the chromatin remodeling complexes help the transcriptional machinery to navigate through tightly packaged DNA–protein complexes known as chromatin.While the basic molecular components of the transcriptional machinery have been well characterized, the mechanisms by which they orchestrate distinct patterns of gene expression are only now being illuminated. Studies suggest that various components of the machinery may assemble in a combinatorial manner that is dependent on the status of the cell. However, the specific assemblies that arise at a given time or place, and the signals that trigger their assembly and disassembly, are not well understood. Recent studies suggest that covalent modifications may play critical roles in various stages of the transcriptional process, thereby facilitating specificity and temporal control.
Gene-specific transcription: posttranslational modification of transcription factors
Protein phosphorylation is widely recognized as a common mechanism for regulating transcription factors, and this mechanism is beautifully conserved throughout evolution (reviewed in ref. 3). In addition to phosphorylation, however, transcription factors and coactivators are subject to a host of other PTMs. Here, we consider the impact of PTMs in the context of two case studies: CREB (cAMP-responsive element binding protein) and p53.Regulation of CREB by PTMs
CREB, the first transcription factor shown to be modified by phosphorylation, plays a central role in the regulation of glucose homeostasis, growth-factor-dependent cell survival and memory storage.4 Addition of a phosphoryl group to a single site, Ser133, activates CREB-dependent transcription by promoting the association of CREB with the coactivator CBP (CREB binding protein or its relative p300; Fig. 2). NMR studies have shown that the kinase-inducible domain of CREB undergoes a structural transition from random-coil to alpha-helix upon phosphorylation and CBP interaction (Fig. 3). Once assembled, the CREB–CBP complex is believed to stimulate gene expression through the recruitment of an active RNAP II complex and the remodeling of chromatin via histone acetylation. Thus, phosphorylation of Ser133 serves as a key molecular switch to activate CREB. | ||
| Fig. 2 Domain structure of CREB. Phosphorylation at Ser133 activates CREB-dependent transcription by recruiting the coactivator CBP/p300. Q1 and Q2, glutamine-rich domains; KID, kinase-inducible domain; bZIP, DNA-binding leucine zipper domain. | ||
 | ||
| Fig. 3 NMR structure of the complex of the kinase-inducible domain of CREB (yellow) with the KIX domain of CBP (cyan). Ser133 is located at the amino terminus of helix αB and forms an ion pair with Lys652 and a hydrogen bond with Tyr658 of CBP. Together, these two contacts account for nearly half the free energy of complex formation. (Copyright permission from: Nat. Rev. Mol. Cell Biol.http://www.nature.com/) | ||
Intriguingly, a wide variety of stimuli, including cyclic AMP, phosphoinositol, nerve growth factor, and KCl-induced depolarization of neurons, leads to phosphorylation of Ser133 with comparable stoichiometry and kinetics. Consistent with this observation, CREB is a substrate for numerous protein kinases, including 90 kDa ribosomal S6 kinase (pp90RSK), protein kinase C (PKC), mitogen- and stress-activated protein kinase (MSK-1), mitogen-activated protein kinase-activated protein kinase-2 (MAPKAP-2) and Ca2+, calmodulin-dependent protein kinases (CaMK) II and IV. Efforts to understand how CREB distinguishes between distinct cellular inputs have led to the discovery of additional covalent modifications that impact CREB activity.
Functional roles for two other phosphorylation sites, Ser142 and Ser143, have been uncovered.4–6 Phosphorylation at Ser142 by CaMKII prevents formation of the CREB–CBP complex in vitro and blocks the activation of target genes in transfected mammalian cells. On the other hand, Ser142 phosphorylation has also been linked to the positive regulation of CREB activity. For instance, Gau et al. demonstrated that both Ser142 and Ser133 participate in resetting of the circadian clock using a mouse mutant lacking the Ser142 phosphorylation site (S142A).5 Down-regulation of the clock-keeping genes, c-fos and mPer1, was observed in the mouse mutant, suggesting that phosphorylation of Ser142 activates CREB-dependent transcription. Studies by Kornhauser et al. lend further support to a stimulatory role for Ser142.6 In particular, the triply phosphorylated form of CREB was required for effective calcium depolarization-induced transcription in neurons. These studies reveal an interesting interplay among the three sites and suggest that distinct combinations of PTMs may be capable of triggering specific programs of gene expression.
In addition to phosphorylation, CREB has been shown to be regulated by O-GlcNAc glycosylation, the covalent modification of serine and threonine residues by β-N-acetylglucosamine (Fig. 4).7 The O-GlcNAc modification is present in all higher eukaryotic organisms from C. elegans to man. It has been shown to be ubiquitous, inducible and highly dynamic, suggesting a regulatory role analogous to phosphorylation.8 Glycosylation of CREB occurs at two sites within the transactivation domain. Addition of the sugar disrupts the interaction between CREB and TAFII130, a component of the transcriptional machinery, and hinders the ability of CREB to activate gene transcription in vitro. Thus, glycosylation may function as a ‘brake’ to repress the activity of CREB and counteract the stimulatory effect of phosphorylation. Consistent with a role for O-GlcNAc in transcriptional repression, the enzyme that catalyzes the modification (O-GlcNAc glycosyltransferase, OGT) has been shown to associate with the transcriptional repressor protein mSin3A and thereby synergistically suppress transcription.8 Elucidating the dynamic interplay between glycosylation and phosphorylation of CREB and the extracellular stimuli that modulate O-GlcNAc glycosylation levels should provide additional insight into the physiological significance of this unusual modification.
Recently, CREB was also shown to undergo acetylation at three lysine residues within the kinase inducible domain.9 While the importance of histone acetylation has been recognized for nearly 30 years, recent studies have revealed much broader roles for this modification than were suspected previously. The addition of an acetyl group has been shown to modulate a number of functional properties of transcription factors. For instance, acetylation can decrease the binding of transcription factors to DNA promoters, attenuate transcription by disrupting transcription factor–coactivator complexes or cause nuclear sequestration of transcription factors. In the case of CREB, mutation of the three acetylated lysine residues to alanine or arginine markedly enhanced CREB-dependent gene expression. While Lu et al. have proposed that acetylation enhances the transactivation potential of CREB, the precise functional impact of acetylation warrants further study given the considerable structural differences between acetylated lysine and the alanine/arginine mutations.
As improved methods for studying PTMs are developed, additional modifications will undoubtedly enter into the arena. Continued investigation into the functional roles of these modifications and the cross-talk between them is expected to provide a deeper understanding of CREB-mediated biological processes such as neuronal survival, glucose homeostasis and memory storage.
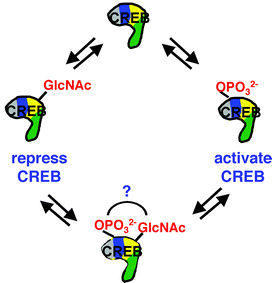 | ||
| Fig. 4 Illustration of the potential interplay between dynamic O-GlcNAc glycosylation and phosphorylation. | ||
Regulation of p53 by PTMs
The transcription factor p53 plays a critical role in suppressing tumor development by initiating a program of cell cycle arrest and programmed cell death (apoptosis) in abnormal or stressed cells.10 Apoptosis can prevent cancerous cells from proliferating, but malignancy is often associated with malfunctioning pro-apoptotic machinery in cells. In fact, half of all human cancers are associated with a mutation within the p53 gene, while many others show deficiency in the p53-mediated pathway.Due to its dramatic consequences for cellular function, the activity of p53 must be tightly regulated. Under conditions of normal cell growth, p53 activity is tightly suppressed, while DNA damaging agents (genotoxic stimuli) lead to rapid induction of p53 to initiate programmed cell death. This intricate regulation involves a plethora of posttranslational modifications, which include at least 18 distinct sites in the human form of the protein. Elucidating the precise roles of each modification is beginning to reveal how this important transcription factor responds to diverse environmental cues. Moreover, insights into the mechanisms of p53 regulation are yielding new and exciting drug targets for cancer therapy (reviewed in ref. 11.)
Various toxic stimuli induce both the accumulation and activation of p53, including genotoxic radiation, inhibitors of DNA replication and transcription, as well as osmotic and heat shock (Fig. 5).12 One of the primary ways by which cellular stress is relayed to p53 is via kinase pathways. Indeed, inducible phosphorylation of p53 has been observed at 9 sites within the N-terminal region of the protein and at 2 sites within the C-terminal region, with distinct phosphorylation events mediated by different kinases and induced by particular stimuli. For example, phosphorylation of Ser15 is strongly induced by DNA damaging agents such as UV and γ-radiation, cisplatin and camptothecin, but it is not induced in response to the RNA polymerase II inhibitor actinomycin D, which is nonetheless known to activate p53. While both UV and γ-radiation stimulate the rapid phosphorylation of Ser15 within 1–2 h, phosphorylation of Ser 46, in contrast, is significantly delayed.13 The kinase that phosphorylates Ser46 in response to UV radiation (homeodomain-interacting protein kinase 2, HIPK2) cannot phosphorylate Ser46 following γ-radiation, suggesting that these two types of DNA damaging events lead to divergent pathways.14 Interestingly, phosphorylation of Ser 46 by HIPK2 is necessary but not sufficient to induce apoptosis of target cells. Indeed, UV radiation leads to phosphorylation not only of Ser15 and Ser46, but also of Ser6, Ser9, Ser20 and Ser37. Thus, multiple concerted phosphorylation events appear to be required for UV-induced apoptosis.
 | ||
| Fig. 5 Domain structure of p53 and sites of phosphorylation (P), acetylation (Ac) and sumoylation (S) in response to DNA damaging events (e.g. UV and γ-radiation, and the DNA strand breaker camptothecin); cellular stress (e.g. oxygen deprivation mimicked by deferoxamine mesylate); transcriptional inhibition induced by the RNAP II inhibitor actinomycin D. NLS, nuclear localization signal domain; TET, tetramerization domain; REG, regulation of DNA-binding domain. | ||
The mechanisms by which phosphorylation induces p53 accumulation and activity are only beginning to be understood. In many cases, phosphorylation appears to be linked to protein stability.12 In unstressed cells, p53 accumulation is prevented by association of the N-terminus of p53 with the ubiquitin ligase MDM2. MDM2 decorates numerous p53 lysines with the 76-residue protein ubiquitin, a signal that targets p53 for rapid degradation. Several phosphorylation sites within the N-terminus of p53 have been shown to disrupt the specific interaction between p53 and MDM2. For example, phosphorylation of Ser20 appears to be necessary for stabilizing p53 in vivo in response to UV and γ-radiation, and significantly disrupts binding to MDM2 in vitro. Moreover, the kinase that phosphorylates Ser20 in vitro (checkpoint kinase 2) was shown to be required for p53 stabilization in response to γ-radiation. Likewise, phosphorylation of Thr18 directly disrupts the interaction of p53 with MDM2 in vitro.15 Intriguingly, the authors found that phosphorylation at Thr18 in response to DNA damage was ablated in a mutant missing Ser15. Indeed, in vitro attempts to phosphorylate Thr18 were only successful if Ser15 was previously phosphorylated. These data highlight the exciting interplay of PTMs at work in the p53 N-terminus during stabilization and activation.
In contrast to phosphorylation events at the N-terminus, modifications close to the C-terminus influence the transcriptional activity of p53 rather than its stabilization. Many studies suggest that the C-terminal region acts as a negative regulator of p53 transactivation, primarily by inhibiting sequence-specific DNA binding. For instance, phosphorylation of Ser392, which is induced upon genotoxic radiation, activates sequence-specific DNA binding in vitro.10 Similarly, phosphorylation of Ser315 also enhances sequence-specific DNA binding in vitro, and mutation of this site to alanine reduces the transcriptional activity of p53 in vivo.12 Notably, phosphorylation at Ser315 as well as Ser33 and Thr81 promotes binding of p53 to the peptidyl-prolyl isomerase Pin1. The consequence of Pin1 binding is a conformational change in p53 that has a stimulatory effect on p53 activity in vivo.16
In addition to phosphorylation, acetylation also plays a pivotal role in p53 function. Acetylation at multiple lysine residues in the C-terminus is induced in vivo in response to virtually all cellular stresses that activate p53 and is mediated by the acetyltransferase activity of CBP/p300 and the closely-related CBP/p300-associated factor (PCAF) in vitro. Interestingly, acetylation can also be triggered by phosphorylation, suggesting an interplay between the two modifications. Specifically, recent studies indicate that phosphorylation of Ser46 by HIPK2 is necessary for CBP-mediated acetylation at Lys382, which in turn augments p53 activation in response to UV radiation.14 Growing evidence suggests that acetylation, like phosphorylation, may play multiple roles in stabilizing and activating p53. Modification by CBP/p300 and PCAF has been shown to significantly increase p53 sequence-specific binding to DNA in vitro. Additionally, acetylation of specific lysine residues in vitro has been shown to block ubiquitination at those sites as well as nearby non-acetylated lysines.17 Moreover, the same authors show that acetylation significantly extends the half-life of p53 in cells. Finally, recent evidence suggests that the primary role of acetylation may be to activate p53-mediated transcription by recruiting coactivators and histone acetyltransferases to p53-responsive genes.
Acetylation and phosphorylation are only two of the best-characterized modifications that impact p53 function. The transcription factor has been demonstrated to bear numerous other modifications, including ubiquitination, sumoylation (the addition of the small ubiquitin-like protein SUMO), O-GlcNAc glycosylation and ribosylation.10 While these other modifications will not be considered in detail here, the addition of ubiquitin plays a particularly important role in p53 stability.17 Interestingly, an early candidate for a p53-stabilizing drug, CP-31398, identified by Pfizer, functions in vivo by inhibiting the ubiquitination of p53. Although the mechanism by which CP-31398 inhibits ubiquitination is unclear, CP-31398 represents a promising prototype for cancer therapy.18 While many more modifications of p53 remain to be elucidated, it is clear that a complex network of posttranslational modifications permits the fine-tuned regulation of this vital transcription factor.
Unraveling of the DNA: posttranslational modifications of histone proteins
The expression of genes in higher organisms depends critically on the accessibility of the DNA being transcribed. In the nucleus, DNA is packaged around a set of histone proteins (H2A, H2B, H3 and H4), which together with histone H1 and other non-histone proteins, maintain chromatin in its condensed state, inaccessible to the transcriptional machinery.19 In order for transcription to proceed, the condensed chromatin must be unraveled. While the precise mechanisms behind this process are not fully understood, the covalent modification of histones has emerged as a key determinant. Protruding from the nucleosomes of chromatin are the charged N-terminal “tails” of histones. These flexible tails contain several residues that are subject to a diverse array of PTMs, including acetylation, phosphorylation, methylation and ubiquitination. Distinct histone modifications or “marks” have been linked to specific transcriptional states. A set of proteins appears to recognize the marks, thereby dictating dynamic transitions between transcriptionally active or repressed chromatin states. Thus, the presence of a PTM may specify a ‘code’ that determines a distinct regulatory outcome.As histone modifications have been extensively reviewed,19 we highlight here only a few examples. Acetylation of histones H3 and H4 at specific lysine residues has been correlated with transcriptional activity, and accordingly, acetyl groups are selectively recognized by coactivators containing bromo-domains and intrinsic histone acetyltransferase activity (Fig. 6). Consistent with a stimulatory role for many acetyl marks, the histone deacetylases (HDACs) that catalyze the reverse reaction have been linked to transcriptional repression. Although less is known about histone phosphorylation, the addition of a phosphoryl group to Ser10 of histone H3 has emerged as a fundamental player in both transcriptional activation and chromosomal condensation during mitosis. For example, dramatic increases in Ser10 phosphorylation are induced at active loci during the heat-shock response in Drosophila melanogaster. More recently, the discovery of lysine and arginine methyltransferases that are targeted to specific promoters has revealed an exciting regulatory role for histone methylation. Methylation of Lys9 of histone H3 specifically recruits a well-known repressor protein, HP1, to silent heterochromatic sites. On the other hand, methylation of Lys4 has been associated with active genes on euchromatin, and arginine methylation of histone H3 is present only when nuclear receptor-regulated promoters are active. Methylation thus can exert opposing effects on transcriptional regulation, depending on the specific residue involved.
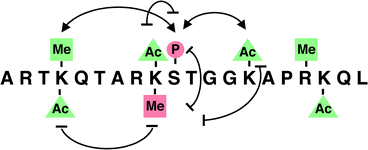 | ||
| Fig. 6 Posttranslational ‘marks’ on the amino terminus of histone H3. Modifications linked to transcriptional activation are shown in green; repressive marks are indicated in red. Potential synergistic interactions are illustrated as connected arrows, while antagonistic interactions are shown as a blocked oval line. P, phosphorylation; Ac, acetylation; Me, methylation. | ||
The story becomes more fascinating when one considers the interplay between various histone modifications and the potential for combinatorial diversity. Specific patterns and temporal sequences of modification events have been observed in vivo, and the presence of a given modification can facilitate or block the presence of a second modification elsewhere on the same histone.19 In the case of histone H3, for example, phosphorylation of Ser10 enhances acetylation of Lys14 by Gcn5 in vitro and precedes Lys14 acetylation at specific promoters in vivo. Furthermore, deacetylation of Lys14 precedes and is required to facilitate Lys9 methylation by the histone methyltransferase Clr4. Other studies have shown that phosphorylation of Ser10 blocks Lys9 methylation in vitro, and conversely, methylation of Lys9 prevents Ser10 phosphorylation both in vitro and in vivo. Thus, specific histone modifications can function to enhance or antagonize one another, providing a mechanism to induce and stabilize specific pairings.
A prediction of the histone code hypothesis is that the sum of the modifications, both in kind and number, will specify distinct biological outcomes, such as gene activation versus gene silencing or more globally, cell proliferation versus cell differentiation. It remains to be seen, however, whether each modification pattern will encode a unique downstream consequence or whether certain modifications will instead tune the specificity, duration or magnitude of the transcriptional response. Nonetheless, the covalent modification of histones clearly encodes functional information essential for chromatin-mediated processes.
RNA polymerase II and the potential CTD code
The transcription of genes is a multi-staged process, which depends on the effective recruitment by transcription factors of coactivators, RNAP II and general transcription factors to target genes. As we have seen, all of the central players in this process require posttranslational control for stability or protein–protein interactions, and RNAP II is no exception. Covalent modifications to the carboxy-terminal “CTD” domain of the largest subunit of RNAP II play an essential role in governing the transcriptional initiation, elongation and termination stages of transcription.2 In mammals, the CTD is composed of 52 repeats of the consensus heptapeptide Tyr-Ser-Pro-Thr-Ser-Pro-Ser. As the transcriptional initiation complex is formed, the CTD remains unphosphorylated (Fig. 7). Partial phosphorylation of the CTD is associated with disruption of RNAP II-promoter interactions (promoter clearance) and the recruitment of enzymes that cap the 5′ end of the nascent RNA. Further phosphorylation promotes transcript elongation and the recruitment of the pre-mRNA splicing machinery. At the final stages of transcription, the CTD recruits protein factors for transcript cleavage and 3′ polyadenylation. Thus, phosphorylation of the CTD plays a critical role during all phases of transcription by assembling a multitude of necessary factors. | ||
| Fig. 7 Changes in the phosphorylation state of the carboxy-terminal domain (CTD) of RNAP II during the initiation and elongation phases of transcription coordinate the transcription process. The initiation phase is associated with phosphorylation of Ser5 of the CTD and the recruitment of capping factors to the nascent RNA transcript. Elongation is associated with additional phosphorylation of the CTD at Ser2 and recruitment of factors involved in elongation and processing of the nascent RNA. O-GlcNAc glycosylation may oppose phosphorylation by preventing transcriptional activation. | ||
But how does the CTD recruit distinct factors during the course of the transcriptional process? One mechanism may involve differential phosphorylation of the CTD tail. Notably, phosphorylation of the CTD dynamically changes over the course of transcription.2 Phosphorylation of Ser5 in the CTD motif is observed between transcription initiation and promoter clearance, whereas phosphorylation of Ser2 occurs only during the elongation phase of transcription. These changes in phosphorylation may recruit distinct factors required for the respective stages of transcription.
Indeed, in yeast, the phosphorylation state of the CTD specifically mediates interactions with a transcription elongation complex (PAF), which in turn coordinates transcriptional regulatory signals and the posttranslational modification of chromatin.20 PAF recruits the yeast Set1 and Set2 histone methyltransferases to the coding regions of DNA. As histone methylation mediated by Set1 is enriched in the 5′ region of transcribed genes, it is speculated that histone tails modified by Set1 may serve as docking sites for various RNA processing complexes during the initiation phase of transcription. In contrast, Set2 appears to function as an elongation factor, with histone tails methylated by Set2 providing potential docking sites for factors involved in later stages of elongation and termination.
Interestingly, the CTD of RNAP II has also been shown to undergo O-GlcNAc glycosylation when present in the unphosphorylated state in vivo.8 Studies suggest that glycosylation may function in opposition to phosphorylation. For instance, synthetic CTD peptides that undergo glycosylation at Thr4 of each CTD repeat cannot be phosphorylated by the TFIIH kinase CDK7.21 Conversely, phosphorylated peptides are not substrates for the O-GlcNAc glycosyltransferase OGT, even though the two primary sites of modification are distinct. A functional relationship between OGT and RNAP II has been further suggested by recent observations that RNAP II and OGT form a complex in vivo.8 One implication of these studies is that glycosylation of RNAP II may prevent transcriptional activation and elongation, consistent with a general role for O-GlcNAc in transcriptional repression (see also discussion of CREB). It will be interesting to establish whether OGT represents a bona-fide transcriptional repressor in this context and to uncover mechanisms for relieving this inhibition.
Future directions
The process of transcription vividly demonstrates the complex and finely tuned role of PTMs in controlling biological processes. Further efforts to uncover novel modification forms, to elucidate the physiological functions of PTMs, and to explore their dynamic regulation in cells should enhance our understanding of this important regulatory manifold.Growing interest in these modifications has spurred the development of exciting methodologies to tackle the study of PTMs.22 For example, multidimensional protein identification technology (MudPIT), which uses multiple chromatographic steps coupled to mass spectrometry, has enabled global studies of posttranslationally modified proteins in yeast. Chemical derivatization methods and metal ion affinity chromatography combined with mass spectrometry have facilitated the enrichment of modified peptides and the sequencing of modification sites. Moreover, complementary approaches such as ‘bump-hole’ type strategies have identified novel physiological substrates of protein kinases.23 Most of these approaches, however, have been most successful when applied to small sets of phosphorylated proteins in specific functional contexts. As such, a major obstacle to the study of PTMs remains the paucity of methods for tracking the wide variety of modifications in vivo. One complication lies in the nature of PTMs themselves – i.e., most modifications are fleeting, present in low cellular abundance, and/or restricted to specific cell types or subcellular compartments. The need for robust methods to detect PTMs rapidly and with requisite sensitivity represents an opportunity for chemists to extend existing strategies and to develop new technologies.
The study of PTMs will likely reveal previously unknown modes for the regulation of protein function. Approaches that allow for the homogeneous incorporation of modified amino acids into proteins will be essential for elucidating the diverse functional roles of PTMs.24 In particular, the incorporation of unnatural amino acids via nonsense suppression and the semi-synthesis of proteins via expressed protein ligation will continue to be invaluable tools for understanding structure–activity relationships. Moreover, the identification of specific modification sites will enable further dissection of the precise functional roles of individual modifications using genetic approaches.
The potential for covalent modifications to be dynamically regulated in response to extracellular signals raises fascinating questions. It will be important to determine which modifications are inducible and to elucidate the enzymes and pathways involved. In contrast to kinase pathways, the signaling cascades leading to other modifications such as acetylation, glycosylation and methylation are less well understood. Will histone demethylases be discovered, for example? What cellular signals will stimulate a particular covalent modification or pattern of modifications? Notably, some PTMs, including deacetylation by the NAD-dependent sirtuin enzymes and O-GlcNAc glycosylation, have been shown to respond directly to metabolic cues. Thus, further insights will be gleaned by investigating PTMs in various physiological or pathological contexts. For such studies, the synthesis of modified peptides and the generation of selective antibodies should prove to be invaluable tools. Just as phospho-specific antibodies have expanded our understanding of kinase pathways, antibodies against other PTMs should enable the levels of specific modifications to be monitored in response to various stimuli. Moreover, advances in quantitative mass spectrometry analysis and approaches that allow modification events to be monitored in living cells via fluorescence microscopy should provide further insight into the dynamic regulation of PTMs.25
Lastly, understanding the interrelationship between multiple different modifications will be critical for unraveling the combinatorial ‘code’ of PTMs. The ability of one modification to antagonize or synergize the deposition of another modification can have important biological consequences, and dynamic transitions among various modification states could serve to regulate the spatial and temporal behavior of proteins. At present, it remains to be seen what fraction of the protein molecules will experience a given set of modification events. In this regard, the development of new approaches to detect low levels of modifications in cells and to isolate proteins bearing discrete modifications will be essential. Ultimately, efforts to sort out the ‘molecular switchboard’ may lead to novel insights and modes for manipulating gene expression and other important biological processes.
References
- R. G. Krishna and F. Wold, in Advances in Enzymology and Related Areas of Molecular Biology, ed. A. Meister,John Wiley & Sons, Inc., New York, 1993, vol. 67, pp. 265–298 Search PubMed; ; http://www.abrf.org/index.cfm/dm.home?AvgMass=all.
- G. Orphanides and D. Reinberg, Cell, 2002, 108, 439–451 CAS.
- A. H. Brivanlou and J. E. Darnell Jr., Science, 2002, 295, 813–818 CrossRef CAS.
- B. Mayr and M. Montminy, Nat. Rev. Mol. Cell Biol., 2001, 2, 599–609 CrossRef CAS.
- D. Gau, T. Lemberger, C. von Gall, O. Kretz, N. L. Minh, P. Gass, W. Schmid, U. Schibler, H. W. Korf and G. Schutz, Neuron, 2002, 34, 245–252 CAS.
- J. M. Kornhauser, C. W. Cowan, A. J. Shaywitz, R. E. Dolmetsch, E. C. Griffith, L. S. Hu, C. Haddad, Z. Xia and M. E. Greenberg, Neuron, 2002, 34, 221–233 CAS.
- N. Lamarre-Vincent and L. C. Hsieh-Wilson, J. Am. Chem. Soc., 2003, 125, 6612–6613 CrossRef CAS.
- N. E. Zachara and G. W. Hart, Chem. Rev., 2002, 102, 431–438 CrossRef CAS; S. P. N. Iyer and G. W. Hart, Biochemistry, 2003, 42, 2493–2499 CrossRef CAS.
- Q. Lu, A. E. Hutchins, C. M. Doyle, J. R. Lundblad and R. P. S. Kwok, J. Biol. Chem., 2003, 278, 15727–15734 CrossRef CAS.
- D. B. Woods and K. H. Vousden, Exp. Cell Res., 2001, 264, 56–66 CrossRef CAS.
- A. N. Bullock and A. R. Fersht, Nat. Rev. Cancer, 2001, 1, 68–76 CrossRef CAS.
- E. Appella and C. W. Anderson, Eur. J. Biochem., 2001, 268, 2764–2772 CrossRef CAS.
- K. Oda, H. Arakawa, T. Tanaka, K. Matsuda, C. Tanikawa, T. Mori, H. Nishimori, K. Tamai, T. Tokino, Y. Nakamura and Y. Taya, Cell, 2000, 102, 849–862 CAS.
- T. G. Hofmann, A. Moller, H. Sirma, H. Zentgraf, Y. Taya, W. Droge, H. Will and M. L. Schmitz, Nat. Cell Biol., 2002, 4, 1–10 CrossRef CAS.
- K. Sakaguchi, S. Saito, Y. Higashimoto, S. Roy, C. W. Anderson and E. Appella, J. Biol. Chem., 2000, 275, 9278–9283 CrossRef CAS.
- P. Zacchi, M. Gostissa, T. Uchida, C. Salvagno, F. Avolio, S. Volinia, Z. Ronai, G. Blandino, C. Schneider and G. Del Sal, Nature, 2002, 419, 853–857 CrossRef CAS.
- C. L. Brooks and W. Gu, Curr. Opin. Cell Biol., 2003, 15, 164–171 CrossRef CAS.
- W. Wang, R. Takimoto, F. Rastinejad and W. S. El-Deiry, Mol. Cell. Biol., 2003, 23, 2171–2181 CrossRef CAS.
- S. L. Schreiber and B. E. Bernstein, Cell, 2002, 111, 771–778 CAS; T. Jenuwein and C. D. Allis, Science, 2001, 293, 1074–1080 CrossRef CAS; S. L. Berger, Curr. Opin. Genet. Dev., 2002, 12, 142–148 Search PubMed.
- M. Hampsey and D. Reinberg, Cell, 2003, 113, 429–432 CAS.
- F. I. Comer and G. W. Hart, Biochemistry, 2001, 40, 7845–7852 CrossRef CAS.
- M. P. Washburn, D. Wolters and J. R. Yates, Nat. Biotechnol., 2001, 19, 242–247 CrossRef CAS; S. B. Ficarro, M. L. McCleland, P. T. Stukenberg, D. J. Burke, M. M. Ross, J. Shabanowitz, D. F. Hunt and F. M. White, Nat. Biotechnol., 2002, 20, 301–305 CrossRef CAS; H. L. Zhou, J. D. Watts and R. Aebersold, Nat. Biotechnol., 2001, 19, 375–378 CrossRef CAS; L. Wells, K. Vosseller, R. N. Cole, J. M. Cronshaw, M. J. Matunis and G. W. Hart, Mol. Cell. Proteomics, 2002, 1, 791–804 Search PubMed.
- A. C. Bishop, O. Buzko and K. M. Shokat, Trends Cell Biol., 2001, 11, 167–172 CrossRef CAS.
- R. M. Hofmann and T. W. Muir, Curr. Opin. Biotechnol., 2002, 13, 297–303 CrossRef CAS; D. A. Dougherty, Curr. Opin. Chem. Biol., 2000, 4, 645–652 CrossRef CAS.
- R. Aebersold and M. Mann, Nature, 2003, 422, 198–207 CrossRef CAS; J. Zhang, R. E. Campbell, A. Y. Ting and R. Y. Tsien, Nat. Rev. Mol. Cell Biol., 2002, 3, 906–918 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: frontispiece figure. See http://www.rsc.org/suppdata/ob/b3/b312466e/ |
| This journal is © The Royal Society of Chemistry 2004 |