Atom-efficient electrophilic aromatic nitration by dinitrogen pentoxide catalysed by zirconium(IV) 2,4-pentanedionate
Adrian J.
Hill
a,
Ross W.
Millar
b and
John P. B.
Sandall
a
aDepartment of Chemistry, University of Exeter, Stocker Road, Exeter, UK EX4 4QD
bQinetiQ, MoD Fort Halstead, Sevenoaks, Kent, UK TN14 7BP
First published on 6th November 2003
Abstract
An atom-efficient, non-acidic, catalytic process is described for the nitration of electron deficient arenes such as o-nitrotoluene using a dinitrogen pentoxide–zirconium(IV) 2,4-pentanedionate system in dichloromethane solvent. Kinetic studies showed the nitration process to be first-order with respect to the aromatic substrate and higher than first-order with respect to the catalyst. Addition of the catalyst at ca. 0.1–1 mol% compared with both N2O5 and the organic substrate results in an increase in the first-order rate constant for nitration by a factor of approximately 5000 with a turnover number of at least 500. The orientation of the nitration products (2,4-/2,6-dinitrotoluenes) is consistent with attack of nitronium ion. The apparently high order of reaction with respect to the catalyst suggests a possible heterogeneous process.
Introduction
Commonly used reagents for nitration of aromatic compounds usually involve a nitronium ion precursor such as nitric acid together with a strong acid (e.g. sulfuric) to generate the required nitronium ion.1 Alternative methods include the use of nitronium salts,2 particularly nitronium tetrafluoroborate and bidentate metal nitrates, e.g. titanium tetranitrate3 dissolved in tetrachloromethane. Other metal salts that have been used include zirconium tetranitrate4 and ceric ammonium nitrate.5 Dinitrogen pentoxide has been examined6 as a nitrating agent; complex reaction pathways through nitronium ion, the oxide itself and also a radical path, via nitrogen trioxide, have been identified. Many catalytic systems have been employed, most of them heterogeneous; thus sulfuric acid may be supported on silica gel and is then found to enhance the rate of nitration of e.g. strongly deactivated aromatics such as nitrobenzene.7 Metal nitrates, e.g. of iron(III) or copper(II), supported on montmorillonite clay8 have been shown to be efficaceous agents as have metal ion-exchanged clays.9 These examples of alternative methods have the advantage of not requiring large quantities of strong acids that require subsequent disposal and they are often advocated on environmental grounds.10Two other examples of catalytic nitration, particularly relevant to our current work, involve the use of lanthanide(III), zirconium(IV) or hafnium(IV) triflate salts as catalysts of nitric acid nitration11 and the use of dinitrogen pentoxide catalysed by iron(III) 2,4-pentanedionate.12 The lanthanide triflates catalyse the nitric acid nitration of bromobenzene in a two-phase aqueous–dichloroethane solvent at reflux temperature; the mechanism was argued to involve the formation of nitronium ion, via nitrate capture by the lanthanide ion which in turn generates the necessary proton. The authors11 suggest that in effect the Brønsted acidity of the nitric acid is enhanced by the Lewis acid–base interaction of the lanthanide cation and nitrate anion. The zirconium and hafnium triflates, with their even higher Z/r ratio arising from the lanthanide contraction, also catalyse (24 hours at reflux) the nitration of o-nitrotoluene, a much more electron deficient arene than bromobenzene. A second method, developed by Bak and Smallridge,12 uses dinitrogen pentoxide with iron(III) 2,4-pentanedionate as catalyst. This system appears to be a more powerful nitrating agent than the nitric acid–lanthanide catalyst, giving m-dinitrobenzene from nitrobenzene (94% yield, 4 minutes, 40 °C).
Results and discussion
To investigate catalytic systems of this type further we have carried out a kinetic investigation of a nitration system consisting of dinitrogen pentoxide and zirconium(IV) 2,4-pentanedionate (Zr(acac)4) dissolved in dichloromethane (DCM) using o-nitrotoluene as substrate, since it reacted smoothly at a convenient rate (half-life 2–5 minutes) at 0 °C giving a quantitative yield of 2,4- and 2,6-dinitrobenzenes. A kinetic study was chosen in order to assess quantitatively the catalytic power of the system; simply measuring yields after a certain period of time may be misleading. A series of preliminary experiments was carried out to determine the stoichiometry of the reaction: gradually increasing amounts (0.03 to 0.47 mmol) of the catalyst were dissolved in dried dichloromethane and then 2.50 ± 0.05 mmol of N2O5 was added. After subsequent addition of 1.45 mmol o-nitrotoluene, the amounts of unreacted organic substrate and dinitrotoluenes produced were determined. These experiments showed that 1 mmol of catalyst reacted with/complexed 8.0 ± 0.5 mmol N2O5 to form a species that did not nitrate the o-nitrotoluene; only amounts of N2O5 in excess of this ratio were consumed by the latter. It seems probable that 4 mmol of N2O5 is lost by nitration of the pentanedionate ligand;13 it is possible that another 4 mmol is rendered passive by coordination to the metal ion centre. Attempts to further characterise this species proved fruitless. Since the catalyst is active in small quantities, this consumption of N2O5 is only preparatively significant with substrates rather more unreactive than nitrobenzene when greater amounts of catalyst are required.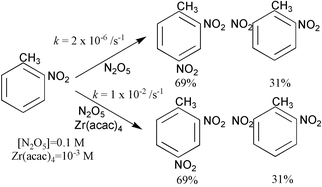
The isomer ratio (2,4-/2,6-dinitrotoluene) was observed to be 69 : 31 (± 1%); very close to the ratio14 for nitric–sulfuric acid mixtures of 67 : 33 where the nitrating agent is agreed to be nitronium ion.
The kinetics of the system were examined (at 0 °C to minimise decomposition of N2O5) by GC analysis of both organic substrate and products. The concentration–time data were fitted to a first-order process independently for both starting material and products; there were no significant differences in fitted rate constants for these components and no deviations from first-order behaviour outside experimental error.
An approximate first-order rate constant for the nitration of o-nitrotoluene in the absence of catalyst was obtained by allowing the reaction to run at 0 °C for 3 hours with an excess of N2O5; an extent of reaction of 5% was observed. A separate experiment showed that nitric acid produced in the reaction did not carry out nitration in the presence of the catalyst.
First-order rate constants are tabulated for runs under various initial conditions (Table 1). For runs 2–6 fresh, non-dried catalyst solutions were used; in runs 7–10, the catalyst solution was dried for the specified time using a molecular sieve. In runs 2–4, the substrate was added last; in runs 5 and 6 it was added before the N2O5. Generally initial starting concentrations of N2O5 were about 0.1–0.3 mol dm−3 and substrate 0.075 mol dm−3. Run 1 (no catalyst) compared with runs 2–4 demonstrates the high activity of this catalytic system; even small quantities enhancing the rate constant by 103–104. Also worthy of note is that the order of addition is significant: if the organic substrate is added to the catalyst solution before the N2O5, activity is reduced by a factor of 5 at typical catalyst loadings: compare runs 2–4 with runs 5 and 6. This implies a lack of perfect homogeneity in the system. Although the catalyst–N2O5–DCM solution appears homogeneous (at the concentrations required), a slight precipitate is observed when the substrate is added; this is slightly different from the behaviour reported12 for the iron(III) 2,4-pentanedionate system where addition of N2O5 to a catalyst solution in DCM gave a pale-pink solid which could not be characterised; this material did not nitrate the substrate but was an effective catalyst when further amounts of N2O5 were added. In this it resembles the behaviour of our catalyst in reacting with/complexing N2O5 to some extent, forming a material which is catalytic only when further N2O5 is added.
| Run | Catalyst added/10−4 M | Rate constant k/10−3 s−1 | Drying time/h |
|---|---|---|---|
| 1 | 0 | 0.002 | 0 |
| 2 | 1.2 | 2.09 | 0 |
| 3 | 2.2 | 9.89 | 0 |
| 4 | 3.2 | 21.5 | 0 |
| 5 | 2.2 | 2.20 | 0 |
| 6 | 3.2 | 5.05 | 0 |
| 7 | 3.0 | 2.76 | 5 |
| 8 | 4.9 | 1.18 | 87 |
| 9 | 3.4 | 0.04 | 111 |
| 10 | 4.5 | 0.06 | 111 |
Traces of water destroy N2O5 and, since the catalyst is hygroscopic, it was dried both before (vacuum oven) and after dissolution in DCM (molecular sieve); however both these processes reduced catalytic activity (runs 7–10) without increasing the amount of available N2O5 for nitration. Some experiments involving o-chloronitrobenzene were also carried out; the much lower reactivity (approximately a factor of 100 compared with the o-nitrotoluene) necessitated the use of much larger amounts of catalyst for the reaction to go at a reasonable rate. Since the active form of the catalyst requires consumption of relatively large amounts of N2O5, precise kinetic studies were not carried out.
It remains to explain the apparent very high order of reaction with respect to the catalyst: compare runs 2, 3 and 4 where a doubling of the initial catalyst concentration leads to an increase in the pseudo first-order rate constant by a factor of 5 and a further 50% increase in catalyst concentration leads to doubling of the constant. This implies that formation of the active catalytic species depends on at least the square of the initial catalyst concentration; the process is clearly more complicated than formation of a simple inner-sphere complex between N2O5 and the zirconium ion. One rather unlikely possibility is that, in the active catalyst species, the N2O5 bridges two or more zirconium centres. However, another more likely possibility is that the catalytic species is in fact the trace of precipitated solid; as the concentration of the zirconium salt increases this could clearly lead to a disproportionate amount of precipitated material when its saturation point is exceeded. This implies a heterogeneous catalytic system, which would also account for the effect of order of addition of substrate and N2O5 mentioned above. The non-crystalline nature of the suspended solid makes its complete characterisation difficult.
Conclusion
Our results show that zirconium 2,4-pentanedionate is an extremely efficient catalyst for N2O5 nitration of aromatic substrates, at least for those of greater reactivity than nitrobenzene. The environmental advantages of N2O5 over acid-based nitration methods are preserved, as are the atom-efficiency and the suitability of the method for aromatics with acid sensitive substituents. The precise mechanism of catalysis requires much further study for its elucidation, but it may involve a solid, or at least colloidal, species pre-formed from the zirconium salt and N2O5. The rate-limiting step of nitration follows, first-order with respect to substrate since the catalyst concentration is effectively constant, in which formation of the nitronium ion from N2O5 is increased relative to that in the absence of catalyst. This probably occurs by the stabilisation of the incipient nitrate ion by the ionic zirconium species. If the system is a truly heterogeneous one then the high efficiency of the catalyst may arise from its high surface area – it appears only as a slight colloidal-type suspension. In this case the reaction takes place on the surface by adsorption of the N2O5. This also explains the observations of Bak and Smallridge.12 The isomer ratio in the product confirms that the nitronium ion is the effective nitrating species. The mechanism differs completely from that of the zirconium triflate–nitric acid system.11Experimental
Materials and methods
o-Chloronitrobenzene (BDH), o-nitrotoluene and pentachloronitrobenzene (Lancaster), 2,4-dinitrotoluene, 2,6-dinitrotoluene (Aldrich) were used without further purification. Dichloromethane (BDH, HPLC grade) was distilled over calcium hydride in silanised glassware. Zirconium(IV) 2,4-pentanedionate (Avocado) was used without purification and also after both oven-drying and drying in DCM solution over molecular sieve. Dinitrogen pentoxide was prepared by the reaction of nitrogen dioxide with ozonised air at room temperature, the gaseous product being passed over phosphorus pentoxide and condensed in acetone–dry-ice cooled ampoules. It was stored at −85 °C until required. All operations involving transfers of chemicals were undertaken in a dry atmosphere glove box. Kinetic runs (dry glove box) were carried out by preparing a solution of Zr(acac)4 (0.02–0.6 mmol) in 20 cm3 DCM, placing the vessel in a liquid paraffin bath thermostatted by a circulating water jacket (0 °C), and adding N2O5 (2.0 mmol) after temperature equilibration. To start the reaction, a solution [5 cm3 of substrate (1.50 mmol)] and a GC internal standard [pentachloronitrobenzene (0.677 mmol)] were finally added. For some o-nitrotoluene reactions, even less catalyst was required and, for accurate measurement, a batch solution of 0.07 g catalyst in 250 cm3 DCM was further diluted as required. Samples for analysis (0.3 cm3) were taken at intervals, quenched in water (10 cm3)–DCM (2 cm3) and analysed by GC. Efficiency of extraction, retention times, and response factors of substrates and products relative to pentachloronitrobenzene were determined separately using authentic materials.Acknowledgements
This work was jointly funded by the EPSRC (Studentship to AJH) and the UK Ministry of Defence as part of the Corporate Research Programme, and the authors gratefully acknowledge financial support from both bodies.References
- G. A. Olah, R. Malhotra and S. C. Narang, Nitration: Methods and Mechanisms, VCH, New York, 1989 Search PubMed.
- (a) D. R. Goddard, E. D. Hughes and C. K. Ingold, J. Chem. Soc., 1950, 2559 RSC; (b) G. A. Olah, S. J. Kuhn and A. J. Mlinko, J. Chem. Soc., 1956, 4257 RSC; S. J. Kuhn and G. A. Olah, J. Am. Chem. Soc., 1961, 83, 4564 CrossRef CAS.
- R. G. Coombes and L. W. Russell, J. Chem. Soc., Perkin Trans. 2, 1974, 830 RSC.
- G. I. Nikishin, E. P. Kaplan and N. I. Kapustina, Izv. Akad. Nauk. SSSR, Ser. Chim., 1976, 1434 Search PubMed.
- S. Dinçtürk and J. H. Ridd, J. Chem. Soc., Perkin Trans. 2, 1982, 961 RSC.
- (a) V. Gold, E. D. Hughes, C. K. Ingold and G. H. Williams, J. Chem. Soc., 1950, 2452 RSC; (b) A. I. Titov, Tetrahedron Lett., 1963, 19, 557 CrossRef CAS.
- N. C. Marziano, C. Tortato, L. Ronchin, F. Martini and C. Bianchi, Catal. Lett., 1999, 58, 81 CrossRef CAS.
- (a) A. Cornélis, A. Gerstmans and P. Laszlo, Chem. Lett., 1988, 1839 CAS; (b) B. Gigante, P. Laszlo, A. Cornélis and M. J. Marcelo-Curto, European Patent 94/19310, 1994.
- B. M. Choudary, M. Sateesh. M. Lakshmi. K. Kotswara Rao, K. V. Ram, K. V. Raghavan and J. A. R. P. Sarma, Chem. Commun., 2000, 25 RSC.
- (Ed.) J. H. Clark, Chemistry of Waste Minimisation, Chapman and Hall, London, 1995 Search PubMed; P. Laszlo, in (Ed.) K. Smith, Solid Supports and Catalysts in Organic Synthesis, Ellis Horwood, Chichester, 1992, Chap. 11 Search PubMed.
- F. J. Waller, A. G. M. Barrett, D. C. Braddock, R. M. McKinnell and D. Ramprasad, J. Chem. Soc., Perkin Trans. 1, 1999, 867 RSC.
- R. R. Bak and A. J. Smallridge, Tetrahedron Lett., 2001, 42, 6767 CrossRef CAS.
- J. P. Collman and W. L. Young, in (Ed.) J. Kleinberg, Inorganic Syntheses, Vol. 7, p. 205, McGraw-Hill Book Company Inc., New York, 1963 Search PubMed.
- J. G. Tillett, J. Chem. Soc., 1962, 5142 RSC.
| This journal is © The Royal Society of Chemistry 2004 |