The fluorine-containing π-allylmetal complex. The transition metal-catalyzed allylic substitution reaction of fluorinated allyl mesylates with various carbon nucleophiles
Tsutomu Konno*, Tsuyoshi Takehana, Takashi Ishihara and Hiroki Yamanaka
Department of Chemistry and Materials Technology, Kyoto Institute of Technology, Matsugasaki, Sakyo-ku, Kyoto 606-8585, Japan. E-mail: konno@chem.kit.ac.jp; Fax: 81-75-724-7580; Tel: 81-75-724-7573
First published on 17th November 2003
Abstract
The allylic substitution reaction of α-fluoroalkylated allyl mesylates with various carbon nucleophiles in the presence of transition metal catalyst (Pd and Mo) proceeded with high regioselectivity to give the corresponding γ-fluoroalkylated products in excellent yields.
Introduction
Transition metal-catalyzed allylic substitution is one of the most efficient and the most versatile methods for carbon–carbon and carbon–heteroatom bond formation, and hence is the focus of intense synthetic attention.1 To date, considerable effort has been devoted to the development of more efficient reactions catalyzed by various transition metals such as palladium,2 molybdenum,3 tungsten,4 iridium,5 rhodium,6 ruthenium,7etc. As a result, a number of reactions are available today which very often afford high regio- and stereo-selectivity. In contrast to the remarkable progress made for the reaction of non-fluorinated allylic substrates, little attention has been paid to the transition metal-catalyzed allylic substitution reaction of fluorine-containing allylic esters so far. There have been quite limited studies on the allylic substitution reaction in fluorine chemistry.8 Recently, we have reported that the reaction of α-fluoroalkylated allyl mesylates with various carboxylates and amines in the presence of palladium catalyst proceeds in a highly regio- and stereo-selective fashion to give the corresponding γ-products, allylic alcohol or amine derivatives in excellent yields, respectively9 (Scheme 1). | ||
| Scheme 1 | ||
Herein we wish to report an extension of our studies to the allylic substitution reaction of fluorine-containing allylic mesylates with various carbon nucleophiles in detail (Scheme 2).
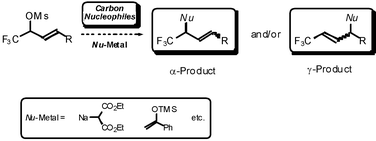 | ||
| Scheme 2 | ||
Results and discussion
We initially examined the reaction of α-trifluoromethylated allyl mesylate 1a with diethyl sodiomalonate 2a using various palladium catalysts as listed in Table 1.10 It was found that the ligand on palladium played an important role in the present allylic substitution reaction. Thus, the use of Pd2(dba)3·CHCl3 resulted in the complete recovery of the starting material. In contrast, the addition of various monodentate ligands such as triphenylphosphine, tri(2-furyl)phosphine, triphenyl phosphite, tributylphosphine, and t-octyl isocyanide was found to facilitate the reaction remarkably (entries 2–10). Additionally, the ratio of palladium and the ligand was crutial for effecting the high yield (entries 2–6). It was found that the palladium complexes which were prepared from palladium(0) species and triphenylphosphine in a ratio of 4 : 1, gave the corresponding allylic substitution product 3a-E in excellent yields (entry 4–6). On the other hand, bulky ligands such as (o-Tol)3P did not provide the desired product at all (entry 8). Changing the ligand on palladium into a bidentate ligand such as dppe, dppf, and bpy caused a large decrease in the formation of 3a-E (entries 9–11). In all cases, neither γ-Z-product 3a-Z nor α-E or Z-products 4a-E, 4a-Z were detected (Fig. 1).11
| Entry | Catalyst | Yield of 3a-E (%)a | Unreacted 1a (%)a |
|---|---|---|---|
| a Determined by 19F NMR. Value in parentheses is of isolated yield.b A diene was produced in 16% yield.c A diene was formed in 8% yield (see Scheme 3). | |||
| 1 | 1/2[Pd2(dba)3·CHCl3] | 0 | 96 |
| 2 | 1/2[Pd2(dba)3·CHCl3] + PPh3 | 38b | 44 |
| 3 | 1/2[Pd2(dba)3·CHCl3] + 2PPh3 | 82c | 0 |
| 4 | 1/2[Pd2(dba)3·CHCl3] + 4PPh3 | 90 | 0 |
| 5 | Pd(OAc)2 + 5PPh3 | 93 | 0 |
| 6 | Pd(PPh3)4 | (99) | 0 |
| 7 | 1/2[Pd2(dba)3·CHCl3] + 4P(2-furyl)3 | 87 | 0 |
| 8 | 1/2[Pd2(dba)3·CHCl3] + 4P(OPh)3 | 61 | 32 |
| 9 | 1/2[Pd2(dba)3·CHCl3] + 4P(n-Bu)3 | 52 | 46 |
| 10 | 1/2[Pd2(dba)3·CHCl3] + 4(t-Oct)NC | 70 | 29 |
| 11 | 1/2[Pd2(dba)3·CHCl3] + 4P(o-Tol)3 | 0 | quant. |
| 12 | 1/2[Pd2(dba)3·CHCl3] + 2dppe | 20 | 75 |
| 13 | 1/2[Pd2(dba)3·CHCl3] + dppf | 0 | 94 |
| 14 | 1/2[Pd2(dba)3·CHCl3] + 2bpy | 19 | 77 |
 | ||
| Fig. 1 | ||
Having identified optimal catalyst, Pd(PPh3)4, a series of nucleophiles were applied to the reaction of 1a and the results are summarized in Table 2.

| Entry | Nucleophile (Nu-Metal) | Temp./°C | Product | Yield of 3 (%)a | Unreacted 1a (%)a |
|---|---|---|---|---|---|
| a Determined by 19F NMR. Values in parentheses are of isolated yields.b An unidentified product was produced in 76% yield.c A diene was formed in 80% yield. | |||||
| 1 | 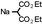 | rt | 3a-E | (99) | 0 |
| 2 |  | rt | 3b-E | (98) | 0 |
| 3 |  | rt | 3c-E | (86) | trace |
| 4 |  | rt | 3d-E | (99) | 0 |
| 5 | 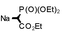 | rt | 3e-E | (94) | 0 |
| 6 |  | rt | — | 0 | 84 |
| 7 |  | 50 | — | 0b | 13 |
| 8 |  | rt | — | 0 | 96 |
| 9 |  | 50 | — | 0 | 79 |
| 10 | PhZnCl | rt | — | 0 | 98 |
| 11 | PhZnCl | 50 | — | 0 | 80 |
| 12 | LiCH2NO2 | rt | — | 0c | 0 |
As shown in entries 1–5, the carbanions derived from diethyl malonate, ethyl acetoacetate, malononitrile, ethyl cyanoacetate, and Hornor–Wadsworth–Emmons reagent reacted smoothly with allyl mesylate 1a in the presence of Pd(PPh3)4 to give the corresponding 3a-E in almost quantitative yields. In all cases, the γ-products with E configuration at the newly created olefinic bond were formed exclusively. No trace of the α-products were detected at all. In entries 2, 4, and 5, the mixture of diastereomers was produced in a ratio of ca. 1 : 1.
We also examined the reaction of allyl mesylate 1a with other carbon nucleophiles such as silyl enol ether, zinc acetylide, phenyl zinc reagent, and lithionitromethane, in the presence of the palladium catalyst at room temperature or 50 °C (entries 6–12). However, the desired products were not formed and the starting material was recovered in almost all cases. In the reaction with lithionitromethane, the decomposition of the π-allylpalladium complex occurred preferentially to lead to the diene 5a in 80% yield (entry 12 and Scheme 3).
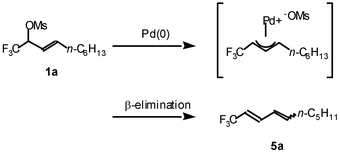 | ||
| Scheme 3 | ||
We next investigated the effect of the side chain R in the allylic mesylate 1 on the present reaction as compiled in Table 3. The substrate 1b having a phenyl group as R was too unstable to isolate, so that the crude material after the mesylation of the corresponding allylic alcohol was employed for the reaction. In this case, a slight decrease in the stereoselectivity at the newly created olefinic bond was observed (entry 2). Changing the substituent from the phenyl to benzyloxymethyl group led to a further decrease in the stereoselectivity at the double bond (entry 3).

Interestingly, in the reaction of 1d where R was H, the bisallylated products 6-EE, 6-EZ, and 6-ZZ were obtained randomly, in addition to the monoallylated products 3h-E and 3h-Z (Table 4, entry 1). In order to obtain the allylic substitution products stereoselectively, we re-examined the effect of transition metal catalysts, as summarized in Table 4.

| Entry | Catalyst | Yield of 3 and 6 (%)a | 3 (E : Z) : 6 (EE : EZ : ZZ)a | Unreacted 1d (%)a |
|---|---|---|---|---|
| a Determined by 19F NMR and GC.b bpy: 2,2′-bipyridyl, 7.c IP(Ph): iminophosphine, 8. IP(Cy): iminophosphine, 9.d Stirred for 24 h.e DME was used as a solvent.f 1,4-Dioxane was used as a solvent.g phen : 1,10-phenanthroline, 10.h DI : diimine, 11.i BPA : bis-picolinylamide, 12. | ||||
| 1 | Pd(PPh3)4 | 95 | 56 (60 : 40) : 44 (43 : 45 : 12) | 0 |
| 2 | 1/2[Pd2(dba)3·CHCl3] | 64 | 39 (100 : 0) : 61 (95 : 5 : 0) | 33 |
| 3 | 1/2[Pd2(dba)3·CHCl3] + 4(o-Tol)3P | 68 | 57 (90 : 10) : 43 (90 : 10 : 0) | 28 |
| 4 | 1/2[Pd2(dba)3·CHCl3] + 2dppe | 85 | 56 (100 : 0) : 44 (95 : 5 : 0) | 13 |
| 5 | 1/2[Pd2(dba)3·CHCl3] + 2bpyb | 85 | 56 (100 : 0) : 44 (97 : 3 : 0) | 5 |
| 6 | 1/2[Pd2(dba)3·CHCl3] + 2IP(Ph)c | 95 | 52 (100 : 0) : 48 (62 : 34 : 4) | 0 |
| 7 | Mo(CO)3(C7H8) | 0 | — | 88 |
| 8 | Mo(CO)3(C7H8) + 2PPh3 | 0 | — | 96 |
| 9 | Mo(CO)3(C7H8) + dppe | 71 | 86 (57 : 43) : 14 (50 : 40 : 10) | 24 |
| 10 | Mo(CO)3(C7H8) + IP(Ph)c | 22 | 96 (77 : 23) : 4 (100 : 0 : 0) | 67 |
| 11 | Mo(CO)3(C7H8) + IP(Cy)c | 18 | 100 (67 : 33) : 0 | 72 |
| 12 | Mo(CO)3(C7H8) + bpy | 29 | 93 (100 : 0) : 7 (100 : 0 : 0) | 66 |
| 13d | Mo(CO)3(C7H8) + bpy | 42 | 90 (100 : 0) : 10 (100 : 0 : 0) | 58 |
| 14e | Mo(CO)3(C7H8) + bpy | 41 | 91 (100 : 0) : 9 (100 : 0 : 0) | 53 |
| 15f | Mo(CO)3(C7H8) + bpy | 92 | 77 (100 : 0) : 23 (100 : 0 : 0) | 4 |
| 16d | Mo(CH3CN)3(C7H8) + bpy | 57 | 84 (100 : 0) : 16 (100 : 0 : 0) | 34 |
| 17 | Mo(CO)3(C7H8) + pheng | 51 | 88 (100 : 0) : 12 (100 : 0 : 0) | 49 |
| 18 | Mo(CO)3(C7H8) + pheng | 54 | 87 (100 : 0) : 13 (100 : 0 : 0) | 37 |
| 19 | Mo(CO)3(C7H8) + DIh | 0 | — | 80 |
| 20 | Mo(CO)3(C7H8) + BPAi | 0 | — | quant. |
Palladium catalysts were employed bearing a variety of ligands such as (o-Tol)3P, dppe, 2,2′-bipyridyl 7, and iminophosphine 8 (shown in Fig. 2). As shown in entries 2–6, the allylic substitution reaction of 1d with 2a proceeded smoothly to give the corresponding products 3 and 6 in good to excellent yields. In all cases, the γ-product was obtained exclusively and no trace of the α-product was detected. However, the monoallylated- and bisallylated products were obtained in almost 1 : 1 ratio and high E selectivity was obtained at the newly formed olefinic bond.
 | ||
| Fig. 2 | ||
In search for more efficient catalysts affording high levels of regio- and stereo-selectivity, we tried using molybdenum catalysts for the allylic substitution reaction of 1d. As shown in entry 7, molybdenum(cycroheptatriene)tricarbonyl did not give the desired product at all. Even when triphenylphosphine was employed as the ligand, in sharp contrast to the palladium catalyst, the product was not formed (entry 8). The use of dppe and iminophosphines (IP(Ph) 8 and IP(Cy) 9) led to the preferential formation of 3, but the stereoselectivity in 3 or the yield was greatly decreased (entries 9–11).
The use of bipyridyl ligand, on the other hand, gave exclusive E-stereoselection at the newly formed olefinic bond as shown in entry 12, although the yield was only 29%. Prolonged reaction time increased the yield from 29 to 42% (entry 13). The reaction in DME did not afford a dramatic change in the yield (entry 14). Of much interest is that the employment of 1,4-dioxane as the solvent improved the reaction markedly, giving a high yield (92%) as well as the high regio- and stereo-selectivity (3 : 6 = 77 : 23, the exclusive E selectivity, entry 15). Phenanthroline 10 was also a good ligand, which afforded high regio- and stereo-selectivity, although the reaction was slightly slow (entries 17 and 18). It was also found that neither diimine 11 nor bispicolinylamide 12 gave the allylated product (entries 19 and 20). In all cases, only γ-products were obtained and no trace of the α-products was detected.
Mechanism
The following mechanism might be proposed, as described in Schemes 4 and 5. | ||
| Scheme 4 | ||
 | ||
| Scheme 5 | ||
Generally, palladium-catalyzed allylic substitution reaction with stabilized carbon nucleophiles occurs via anti oxidative addition-anti nucleophilic attack mode, providing the allylated products with overall retention of configuration (Scheme 4, Path A).12 The molybdenum-catalyzed reaction has also been known to proceed with overall retention of configuration.13 However it has been recently reported that the mode of action of molybdenum catalyst may differ from that of palladium (Path B). Thus, the molybdenum-catalyzed reaction is suggested to take place via syn oxidative addition-syn nucleophilic attack mode.14 Based on this proposed mechanism, the molybdenum complex could coordinate with the allylic mesylate leaving group and an olefin, providing the oxidative product Int-B with the retention of configuration. The nucleophile attacks on the same face as that occupied by Mo, resulting in a net retention in this pathway.
In Int-A or Int-B, the transition metal might be closer to a CF3 group than the R group due to the electron-withdrawing effect of the CF3 group.15 Therefore, the nucleophile reacts preferentially at the less hindered γ-carbon to give the γ-product 3.
In the reaction of 1d (R = H) with sodiomalonate, it is highly possible that sodiomalonate abstracts α-proton of γ-product 3, forming the α-allylated sodiomalonate 13 (Scheme 5). The newly formed nucleophile 13 attacks the γ-carbon of Int-A or Int-B. In this case, Int-A may react more smoothly than Int-B does because of the large steric repulsion between molybdenum species and the bulky nucleophile 13. As a result, higher selectivity of 3/6 was observed in the molybdenum-catalyzed reaction than in the palladium reaction.
For the molybdenum-catalyzed allylic substitution reaction, it has been also reported thus far that the reaction with stabilized carbon nucleophiles proceeds via anti oxidative addition-anti nucleophilic attack mode, like the palladium-catalyzed reaction.16 Therefore, the detailed reaction mechanism for the present study remains unclear at present and an exact explanation for the mechanism awaits further investigation.
Conclusions
In summary, we have investigated the transition metal-catalyzed allylic substitution reaction of α-trifluoromethylated allyl mesylate with various carbon nucleophiles in detail. Only stabilized carbanions such as sodiomalonate, ethyl sodiocyanoacetate, etc. reacted with the mesylate smoothly to give the corresponding alkylation products in excellent yields. It was found that the side chain R in the mesylates affected the reaction significantly. Thus, the monoallylated product was obtained in high yields via palladium-catalyzed reaction in the case of R = n-C6H13, Ph etc., however, the use of 1d (R = H) led to the formation of bisallylated product along with the monoallylated one. The best yield and the best stereoselectivity were obtained when the molybdenum catalyst with a 2,2′-bipyridyl ligand was employed.Experimental
General methods
Infrared spectra (IR) were taken on a Shimadzu FTIR-8200(PC) spectrometer as film on a NaCl plate. 1H and 13C NMR spectra were measured with a Bruker DRX-500 NMR spectrometer in a chloroform-d (CDCl3) solution with tetramethylsilane (Me4Si) as an internal reference. A JEOL JNM-EX90A (84.21 MHz) FT-NMR spectrometer was used for determining 19F NMR spectra in a CDCl3 solution with trichlorofluoromethane as internal standard. High-resolution mass spectra (HRMS) were taken on a Hitachi M-80B mass spectrometer by electron impact (EI), chemical ionization (CI), and fast atom bombardment (FAB) methods. Thin-layer chromatography (TLC) was done on aluminium sheets coated with silica gel (Merck 60 F254), and column chromatography was carried out using silica gel (Wacogel C-200) as adsorbent.Preparation of substrate 1d
To a THF solution of trifluoroacetaldehyde, prepared readily from trifluoroacetaldehyde monohydrate (40 mmol) and conc. H2SO4 (40 mmol), was added a THF solution of vinylmagnesium bromide (20 mmol) at −78 °C. The reaction mixture was stirred for several hours at that temperature, and the reaction was quenched with sat. NH4Cl aq.. The mixture was extracted with ether three times, and the combined organic layers were washed with brine, dried over anhydrous Na2SO4, and filtered. The filtrate was gradually concentrated to remove solvents (THF and ether) to the yellow residue which was used without further purification.To a CH2Cl2 solution of trifluoromethylated allylic alcohol (1 mmol) was added methanesulfonyl chloride (1.2 mmol) and Et3N (1.2 mmol) at 0 °C. The whole was stirred for several hours at that temperature. After quenching the reaction with sat. NH4Cl, the whole was extracted with CH2Cl2 three times, and the combined organic layers were dried over anhydrous Na2SO4, concentrated in vacuo. The residue was purified by silica gel column chromatography to give the corresponding allyl mesylate 1d (∼41% yield).
1-(Trifluoromethyl)-2-propenyl methanesulfonate (1d)
1H NMR (CDCl3) δ 3.11 (3H, s), 5.29 (1H, dq, J = 6.5 Hz, 6.5 Hz), 5.67 (1H, d, J = 10.5 Hz), 5.72 (1H, d, J = 17.2 Hz), 5.91 (1H, ddd, J = 6.5 Hz, 10.5 Hz, 17.2 Hz); 19F NMR (CDCl3) δ − 77.22 (3F, d, J = 6.5 Hz); 13C NMR (CDCl3) δ 39.4, 77.1 (q, J = 34.4 Hz), 122.1 (q, J = 280.9 Hz), 125.3, 126.1; IR (neat) ν 725 (m), 764 (m), 843 (s), 870 (m), 949 (s), 1011 (s), 1109 (m), 1132 (s), 1182 (s), 1271 (s), 1337 (s), 1371 (s), 1418 (m), 2947 (w), 3038 (w); HRMS (CI) m/z 205.0146, found 205.0139 (M + H), calcd for C5H8F3O332S.Typical procedure for the reaction of allyl mesylate 1a with sodiomalonate
To a solution of Pd(PPh3)4 (19 mg, 5 mol%, 0.02 mmol) in THF (3 mL) was added allyl mesylate 1a (93 mg, 0.34 mmol) at 0 °C. After stirring of the reaction mixture for 10 min, sodiomalonate, 2a, prepared from diethyl malonate (1.2 eq. 0.41 mmol) and NaH (1.2 eq. 0.41 mmol), was added to the reaction mixture, followed by warming of the solution to room temperature, then stirred for 2 h. The reaction was quenched with water, and the whole was extracted with ethyl acetate three times. The combined organic layers were washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography to give the corresponding allylated product 3a-E (119 mg, 99% yield).Diethyl 1-(3,3,3-trifluoro-(1E)-propenyl)heptylmalonate (3a-E)
1H NMR (CDCl3) δ 0.87 (3H, t, J = 7.0 Hz), 1.23–1.43 (16H, m), 2.87 (1H, ddt, J = 3.5 Hz, 9.0 Hz, 9.0 Hz), 3.36 (1H, d, J = 9.0 Hz), 4.16 (2H, d, J = 7.5 Hz), 4.20 (2H, d, J = 7.5 Hz), 5.68 (1H, dq, J = 6.5 Hz, 15.5 Hz), 6.27 (1H, ddq, J = 2.0 Hz, 9.0 Hz, 15.5 Hz); 19F NMR (CDCl3) δ −64.74 (3F, d, J = 6.5 Hz); 13C NMR (CDCl3) δ 13.9, 14.0, 22.5, 26.8, 28.9, 31.5, 31.9, 41.8, 56.0, 61.5, 61.6, 120.8 (q, J = 33.5 Hz), 122.6 (q, J = 269.2 Hz), 140.0 (q, J = 6.5 Hz), 167.6, 167.7; IR (neat) ν 671 (m), 795 (s), 856 (m), 979 (m), 1033 (m), 1126 (s), 1280 (s), 1369 (m), 1465 (m), 1681 (m), 1735 (s), 2858 (m), 2931 (s); HRMS (CI) m/z 353.1940, found 353.1938 (M + H), calcd for C17H28F3O4.1-(3,3,3-Trifluoro-(1E)-propenyl)heptylmalononitrile (3c-E)
1H NMR (CDCl3) δ 0.89 (3H, t, J = 7.0 Hz), 1.26–1.35 (8H, m), 1.66–1.69 (1H, m), 1.76–1.78 (1H, m), 2.77 (1H, ddt, J = 5.5 Hz, 9.5 Hz, 9.5 Hz), 3.79 (1H, d, J = 5.5 Hz), 5.96 (1H, dq, J = 6.0 Hz, 16.0 Hz), 6.24 (1H, ddq J = 1.5 Hz, 9.5 Hz, 16.0 Hz); 19F NMR (CDCl3) δ −65.23 (3F, d, J = 6.0 Hz); 13C NMR (CDCl3) δ 13.9, 22.4, 26.5, 27.8, 28.7, 31.4, 43.0, 110.7, 110.9, 121.8 (q, J = 270.1 Hz), 124.7 (q, J = 34.8 Hz), 135.3 (q, J = 6.2 Hz); IR (neat) ν 864 (w), 976 (m), 1130 (s), 1211 (w), 1277 (m), 1315 (m), 1366 (w), 1466 (w), 1686 (w), 2862 (m), 2932 (s); HRMS (CI) m/z 259.1422, found 259.1421 (M + H), calcd for C13H18F3N2.Ethyl 1-(3,3,3-trifluoro-(1E)-propenyl)heptylcyanoacetate (diastereomeric ratio = 60 : 40) (3d-E)
1H NMR (CDCl3) δ 0.87–0.90 (3H, m), 1.29–1.38 (13H, m), 2.83–2.89 (1H, m), 3.50 (1H, d, J = 6.3 Hz) (isomer), 3.64 (1H, d, J = 4.5 Hz) (another isomer), 4.23–4.27 (2H, m), 5.75–5.83 (1H, m), 6.22 (1H, ddq, J = 2.0 Hz, 9.6 Hz, 15.6 Hz) (isomer), 6.27 (1H, ddq, J = 2.0 Hz, 9.7 Hz, 15.6 Hz) (another isomer); 19F NMR (CDCl3) δ −64.65 (3F, d, J = 6.6 Hz) (isomer), −64.73 (3F, d, J = 6.6 Hz) (another isomer); 13C NMR (CDCl3) δ 13.9, 22.5, 26.6, 26.7, 28.7, 28.8, 28.8, 31.0, 31.5, 32.1, 42.3, 42.3, 43.3, 42.4, 63.1, 114.2, 120.3 (q, J = 38.3 Hz) (isomer), 120.5 (q, J = 269.3 Hz) (isomer), 122.7 (q, J = 269.9 Hz) (another isomer), 137.3 (q, J = 6.4 Hz) (isomer), 138.1 (q, J = 6.0 Hz) (another isomer), 164.6, 164.74; IR (neat) ν 856 (w), 980 (m), 1026 (m), 1126 (s), 1277 (m), 1369 (m), 1466 (m), 1682 (m), 1747 (s), 2341 (m), 2862 (m), 2932 (s); HRMS (CI) m/z 306.1681, found 306.1685 (M + H), calcd for C15H23F3NO2.Ethyl 1-(3,3,3-trifluoro-(1E)-propenyl)heptyldiethylphosphonoacetate (diastereomeric ratio = 56 : 44) (3e-E)
1H NMR (CDCl3) δ 0.85–0.88 (3H, m), 1.20–1.47 (19H, m), 2.78–2.89 (1H, m), 2.96 (1H, dd, J = 9.8 Hz, 20.3 Hz) (isomer), 3.04 (1H, dd, J = 7.5 Hz, 21.5 Hz) (another isomer), 4.06–4.19 (4H, m), 4.23 (2H, q, J = 7.0 Hz) (isomer), 5.68 (1H, dq, J = 6.5 Hz, 15.5 Hz) (another isomer), 5.72 (1H, dq, J = 6.5 Hz, 15.5 Hz) (isomer), 6.17 (1H, ddq, J = 2.0 Hz, 10.0 Hz, 15.5 Hz) (isomer), 6.36 (1H, ddq, J = 2.0 Hz, 10.0 Hz, 15.5 Hz) (another isomer); 19F NMR (CDCl3) δ −64.66 (3F, d, J = 6.5 Hz); 13C NMR (CDCl3) δ 13.9, 14.0, 14.1, 16.1–16.3 (m), 22.47, 22.48, 26.4, 26.7, 28.8, 31.5, 31.5, 32.31, 32.37, 41.0 (d, J = 3.4 Hz) (isomer), 41.1 (d, J = 4.1 Hz) (another isomer), 49.7 (d, J = 139.9 Hz) (isomer), 50.7 (d, J = 133.9 Hz) (another isomer), 61.4, 61.6, 62.5 (d J = 7.1 Hz) (isomer), 62.7 (d J = 7.4 Hz) (another isomer), 120.3 (q, J = 33.5 Hz) (isomer), 120.4 (q, J = 33.3 Hz) (another isomer), 122.7 (q, J = 269.2 Hz) (isomer), 140.2–140.5 (m), 167.8 (q, J = 3.9 Hz), 168.2 (q, J = 3.3 Hz); IR (neat) ν 675 (w), 795 (w), 860 (w), 972 (m), 1026 (s), 1123 (s), 1258 (s), 1369 (m), 1466 (m), 1682 (m), 1736 (s), 2858 (m), 2932 (w); HRMS (CI) m/z 417.2018, found 417.2022 (M + H), calcd for C18H33F30O5P.Ethyl 1-(3,3,3-trifluoro-(1E)-propenyl)heptylacetoacetate (diastereomeric ratio = 58 : 42) (3b-E)
1H NMR (CDCl3) δ 0.86 (3H, t, J = 7.0 Hz), 1.22–1.45 (13H, m), 2.17 (3H, s) (isomer), 2.23 (3H, s) (another isomer), 2.90–2.92 (1H, m), 3.46 (1H, d, J = 9.0 Hz) (isomer), 3.50 (1H, d, J = 9.0 Hz) (another isomer), 4.14 (2H, q, J = 7.0 Hz) (isomer), 4.20 (2H, d, J = 7.0 Hz) (another isomer), 5.66 (1H, dq, J = 6.5 Hz, 16.0 Hz) (isomer), 5.67 (1H, dq, J = 6.5 Hz, 16.0 Hz) (another isomer), 6.14–6.21 (1H, m); 19F NMR (CDCl3) δ −64.72 (3F, d, J = 6.5 Hz); 13C NMR (CDCl3) δ 13.95, 14.02, 22.5, 26.8, 26.9, 28.9, 29.6, 30.0, 31.5, 31.6, 32.0, 41.3, 61.6, 61.7, 63.6, 63.8, 120.9 (q, J = 33.6 Hz) (isomer), 121.0 (q, J = 33.6 Hz), 122.5 (q, J = 269.5 Hz) (isomer), 122.6 (q, J = 269.5 Hz) (another isomer), 139.9 (q, J = 6.8 Hz) (isomer), 140.1 (q, J = 6.6 Hz) (another isomer), 167.9, 168.0, 201.1, 201.2; IR (neat) ν 679 (w), 810 (w), 856 (w), 980 (m), 1026 (m), 1126 (s), 1277 (s), 1362 (s), 1466 (m), 1632 (w), 1682 (m), 1720 (s), 2858 (m), 2932 (s); HRMS (EI) m/z 323.1834, found 323.18434 (M + H), calcd for C16H26F3O3.Diethyl 1-benzyloxymethyl-4,4,4-trifluoro-(2E)-butenyl malonate (3f-E)
1H NMR (CDCl3) δ 1.23 (6H, t, J = 7.0 Hz), 3.18–3.23 (1H, m), 3.53–3.64 (2H, m), 3.72 (1H, d, J = 8.0 Hz), 4.10–4.23 (4H, m), 4.45–4.83 (2H, m), 5.67–5.78 (1H, m), 6.47 (1H, ddq, J = 2.0 Hz, 9.5 Hz, 16.0 Hz), 7.28–7.30 (3H, m), 7.33–7.36 (2H, m); 19F NMR (CDCl3) δ −64.95 (3F, d, J = 6.6 Hz); 13C NMR (CDCl3) δ 13.96, 13.97, 41.7, 52.4, 61.6, 61.7, 69.8, 73.2, 121.3 (q, J = 269.2 Hz), 127.6, 127.8, 128.4, 137.5 (q J = 6.5 Hz), 137.7, 167.68, 167.72; IR (neat) ν 700 (m), 739 (m), 864 (w), 978 (m), 1030 (s), 1121 (s), 1369 (s), 1454 (m), 1680 (m), 1732 (s), 2870 (m), 2986 (m); HRMS (FAB) m/z 389.1576, found 389.1580 (M + H), calcd for C19H24F3O5.Diethyl 1-benzyloxymethyl-4,4,4-trifluoro-(2Z)-butenyl malonate (3f-Z)
1H NMR (CDCl3) δ 1.23 (3H, t, J = 7.0 Hz), 3.18–3.23 (1H, m), 3.53–3.64 (2H, m), 3.76 (1H, d, J = 8.0 Hz), 4.10–4.23 (4H, m), 4.45–4.83 (2H, m), 5.67–5.78 (1H, m), 6.23 (1H, dd, J = 11.5 Hz, 11.5 Hz), 7.28–7.30 (3H, m), 7.33–7.36 (2H, m); 19F NMR (CDCl3) δ −58.69 (3F, d, J = 7.8 Hz); 13C NMR (CDCl3) δ 13.97, 14.03, 38.6, 52.2, 61.51, 61.53, 70.4, 73.1, 120.5 (q, J = 33.8 Hz), 122.6 (q, J = 269.2 Hz), 127.6, 127.7, 128.3, 137.8, 139.4 (q J = 6.5 Hz), 167.68, 167.72.Diethyl 4,4,4-trifluoro-1-phenyl-(2E)-butenyl malonate (3g-E)
1H NMR (CDCl3) δ 1.00 (3H, t, J = 7.0 Hz), 1.27 (3H, t, J = 7.0 Hz), 3.84 (1H, d, J = 10.5 Hz), 3.97 (2H, dq, J = 2.5 Hz, 7.0 Hz), 4.18–4.24 (3H, m), 5.65 (1H, dq, J = 6.5 Hz, 15.6 Hz), 6.56 (1H, ddq, J = 2.0 Hz, 8.0 Hz, 15.6 Hz), 7.21–7.23 (1H, m), 7.25–7.28 (1H, m), 7.32–7.34 (2H, m); 19F NMR (CDCl3) δ −64.78 (3F, d, J = 6.5 Hz); 13C NMR (CDCl3) δ 13.7, 14.0, 47.3, 56.8, 61.6, 61.9, 120.2 (q, J = 33.8 Hz), 122.6 (q, J = 269.8 Hz), 127.8, 128.1, 128.9, 137.8, 139.4 (q, J = 6.5 Hz), 166.8, 167.3; IR (neat) ν 559 (m), 608 (w), 673 (w), 700 (s), 766 (m), 862 (m), 978 (m), 1032 (s), 1138 (s), 1454 (m), 1497 (m), 1603 (w), 1678 (m), 1732 (m), 2909 (m), 2986 (s); HRMS (FAB) m/z 345.1314, found 345.1317 (M + H), calcd for C17H20F3O4.Diethyl 4,4,4-trifluoro-(2E)-butenyl malonate (3h-E)
1H NMR (CDCl3) δ 1.27 (6H, t, J = 7.0 Hz), 2.73–2.75 (2H, m), 3.46 (1H, t, J = 7.2 Hz), 4.21 (4H, q, J = 7.0 Hz), 5.72 (1H, dq, J = 7.0 Hz, 16.0Hz), 6.35 (1H, dtq, J = 2.0 Hz, 7.0 Hz, 16.0 Hz); 19F NMR (CDCl3) δ −64.95 (3F, d, J = 7.0 Hz); 13C NMR (CDCl3) δ 14.0, 30.4, 50.5, 61.7, 121.1 (q, J = 33.5 Hz), 122.5 (q, J = 269.3 Hz), 136.0 (q, J = 6.6 Hz), 168.2; IR (neat) ν 862 (w), 970 (m), 1034 (m), 1096 (m), 1128 (s), 1279 (s), 1371 (m), 1448 (m), 1682 (m), 1736 (s), 2988 (m); HRMS (CI) m/z 269.1001, found 269.1002 (M + H), calcd for C11H16F3O4.Diethyl bis(4,4,4-trifluoro-(2E)-butenyl)malonate (6-EE)
1H NMR (CDCl3) δ 1.26 (6H, t, J = 7.3 Hz), 2.43 (4H, d, J = 7.5 Hz), 4.22 (4H, q, J = 7.3 Hz), 5.70 (2H, dq, J = 6.5 Hz, 15.8 Hz), 6.28 (2H, dtq, J = 2.0 Hz, 7.5 Hz, 15.5 Hz); 19F NMR (CDCl3) δ −65.07 (6F, d, J = 6.5 Hz); 13C NMR (CDCl3) δ 13.9, 35.6, 56.4, 62.0, 122.3 (q, J = 269.5 Hz), 122.6 (q, J = 33.7 Hz), 134.3 (q, J = 6.5 Hz), 169.5; IR (neat) ν 681 (w), 856 (w), 974 (m), 1032 (m), 1124 (s), 1205 (s), 1275 (s), 1350 (m), 1447 (m), 1682 (m), 1732 (s), 2988 (s); HRMS (FAB) m/z 377.1188, found 377.1182 (M + H), calcd for C15H19F6O4.References
- For a review on catalytic allylic substitutions, see: (a) J. Tsuji, Palladium Reagents and Catalysts, John Wiley, Chichester, 1995 Search PubMed; (b) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS; (c) T. Hachiya, in Catalytic Asymmetric Synthesis, ed. I. Ojima, VCH, New York, 1993, p. 325 Search PubMed; (d) C. G. Frost, J. Howarth and J. M. Williams, Tetrahedron: Asymmetry, 1992, 3, 1089–1122 CrossRef CAS; (e) G. Consiglio and M. Waymouth, Chem. Rev., 1989, 89, 257–276 CrossRef CAS.
- (a) T. Hayashi, M. Kawatsura and Y. Uozumi, J. Am. Chem. Soc., 1998, 120, 1681–1687 CrossRef CAS; (b) M. Nakoji, T. Kanayama, T. Okino and Y. Takemoto, J. Org. Chem., 2002, 67, 7418–7423 CrossRef CAS; (c) T. Hayashi, M. Kawatsura and Y. Uozumi, Chem. Commun., 1997, 561–562 RSC; (d) U. Kazmaier and F. L. Zumpe, Angew. Chem., Int. Ed., 1999, 38, 1468–1470 CrossRef CAS.
- (a) B. M. Trost, S. Hildbrand and K. Dogra, J. Am. Chem. Soc., 1999, 121, 10416–10417 CrossRef CAS; (b) B. M. Trost, K. Dogra, I. Hachiya, T. Emura, D. L. Hughes, S. Krska, R. A. Reamer, M. Palucki, N. Yasuda and P. J. Reider, Angew. Chem., Int. Ed., 2002, 41, 1929–1932 CrossRef CAS; (c) F. Glorious, M. Neuburger and A. Pfaltz, Helv. Chim. Acta, 2001, 84, 3178–3196 CrossRef; (d) F. Glorious and A. Pfaltz, Org. Lett., 1999, 1, 141–144 CrossRef CAS.
- (a) B. M. Trost and M.-H. Hung, J. Am. Chem. Soc., 1983, 105, 7757–7759 CrossRef CAS; (b) R. Prétôt, G. C. Lloyd-Jones and A. Pfaltz, Pure Appl. Chem., 1998, 70, 1035–1040 CAS.
- (a) R. Takeuchi, Synlett, 2002, 1954–1965 CrossRef CAS; (b) R. Takeuchi, N. Ue, K. Tanabe, K. Yamashita and N. Shiga, J. Am. Chem. Soc, 2001, 123, 9525–9534 CrossRef CAS; (c) T. Ohmura and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 15164–15165 CrossRef CAS; (d) B. Bartels and G. Helmchen, Chem. Commun., 1999, 741–742 RSC.
- (a) P. A. Evans and J. E. Robinson, Org. Lett., 1999, 1, 1929–1931 CrossRef CAS; (b) P. A. Evans and L. K. Kennedy, Org. Lett., 2000, 2, 2213–2215 CrossRef CAS; (c) P. A. Evans and L. J. Kennedy, J. Am. Chem. Soc., 2001, 123, 1234–1235 CrossRef CAS; (d) T. Hayashi, A. Okada, T. Suzuki and M. Kawatsura, Org. Lett., 2003, 5, 1713–1715 CrossRef CAS; (e) P. A. Evans and D. K. Leahy, J. Am. Chem. Soc., 2002, 124, 7882–7883 CrossRef CAS.
- (a) T. Kondo, H. Ono, N. Satake, T. Mitsudo and Y. Watanabe, Organometallics, 1985, 14, 1945–1953.
- (a) Y. Hanzawa, S. Ishizawa and Y. Kobayashi, Chem. Pharm. Bull., 1988, 36, 4209–4212 CAS; (b) Y. Hanzawa, S. Ishizuka, H. Ito, Y. Kobayashi and T. Taguchi, J. Chem. Soc., Chem. Commun., 1990, 394–395 Search PubMed; (c) T. Okano, H. Matsubara, T. Kusukawa and M. Fujita, J. Organomet. Chem., 2003, 676, 43–48 CrossRef CAS.
- (a) T. Konno, K. Nagata, T. Ishihara and H. Yamanaka, J. Org. Chem., 2002, 67, 1768–1775 CrossRef CAS; (b) T. Konno, T. Ishihara and H. Yamanaka, Tetrahedron Lett., 2000, 41, 8467–8472 CrossRef CAS.
- The palladium-catalyzed allylic substitution reactions of fluorine-containing allylic acetate, carbonate, phosphonate, and tosylate have been reported previously. See ref. 8.
- The same high regio- and stereo-selectivity have been reported previously. See ref 8.
- (a) B. M. Trost, Acc. Chem. Res., 1980, 13, 385–393 CrossRef CAS; (b) See ref. 1b.
- (a) B. M. Trost and M. Lautens, J. Am. Chem. Soc., 1982, 104, 5543–5545 CrossRef CAS; (b) B. M. Trost and M. Lautens, J. Am. Chem. Soc., 1987, 109, 1469–1478 CrossRef CAS; (c) B. M. Trost and C. A. Merlic, J. Am. Chem. Soc., 1990, 112, 9590–9600 CrossRef CAS.
- (a) A. V. Malkov, I. R. Baxendale, D. J. Mansfield and P. Kocovsky, J. Chem. Soc., Perkin Trans. 1, 2001, 1234–1240 RSC; (b) D. Dvorak, I. Stary and P. Kocovsky, J. Am. Chem. Soc., 1995, 117, 6130–6131 CrossRef CAS; (c) Y. D. Ward, L. A. Villanueva, G. D. Allred and L. S. Liebeskind, J. Am. Chem. Soc., 1996, 118, 897–898 CrossRef CAS; (d) A. Rubio and L. S. Liebeskind, J. Am. Chem. Soc., 1993, 115, 891–901 CrossRef CAS.
- It has been reported that the palladium atom in the η3 intermediate lies closer to the carbon attached to the electron-withdrawing group. See (a) E. Keinan and M. Pereta, J. Org. Chem., 1983, 48, 5302–5309 CrossRef CAS; (b) E. Keinan and Z. Roth, J. Org. Chem., 1983, 48, 1769–1772 CrossRef CAS.
- (a) B. M. Trost and M. Lautens, Tetrahedron, 1987, 43, 4817–4840 CrossRef CAS; (b) D. L. Hughes, M. Palucki, N. Yasuda, R. A. Reamer and P. J. Reider, J. Org. Chem., 2002, 67, 2762–2768 CrossRef CAS; (c) see ref. 3a.
| This journal is © The Royal Society of Chemistry 2004 |