Esterase-assisted accumulation of 3-carboxy-2,2,5,5-tetramethyl-1-pyrrolidinyloxyl into lymphocytes
Joseph P. Y.
Kao
a and
Gerald M.
Rosen
*b
aMedical Biotechnology Center, University of Maryland Biotechnology Institute, and Department of Physiology, University of Maryland School of Medicine, Baltimore, MD 21201, USA
bDepartment of Pharmaceutical Sciences, University of Maryland School of Pharmacy, and Medical Biotechnology Center, University of Maryland Biotechnology Institute, Baltimore, MD 21201, USA
First published on 12th November 2003
Abstract
The intracellular detection of hydroxyl radical (HO˙) through spin trapping/electron paramagnetic resonance (EPR) spectroscopy has been one of the great challenges in studying free radicals in biology. While 5-carboxy-5-methyl-1-pyrroline N-oxide, [3], can specifically spin trap HO˙ in homogeneous solutions, the ionic nature of nitrone [3] at physiologic pH prevents its entry into cells. We hypothesized that conversion of carboxyl-bearing spin probes such as nitrone [3] into an esterase-hydrolyzable labile ester would permit intracellular localization and accumulation of the spin probes. To test the feasibility of such an approach, we prepared the model compound, 3-acetoxymethoxycarbonyl-2,2,5,5-tetramethyl-1-pyrrolidinyloxyl [7]. This ester enabled ready accumulation of spin label to mM levels in lymphocytes. We suggest that its retention within these cells was the result of intracellular hydrolysis to 3-carboxy-2,2,5,5-tetramethyl-1-pyrrolidinyloxyl [6]. Moreover, our studies show that aminoxyl [6] was stable in the intracellular environment. These model studies suggest a viable strategy for detecting intracellular HO˙ by using the acetoxymethyl ester of 5-carboxy-5-methyl-1-pyrroline N-oxide [3].
Introduction
Nearly fifty years ago oxygen-centered free radicals were proposed to be the thread that linked ionizing radiation and aging.1 More recently, with the discovery that enzymes generate superoxide (O2˙−) and that there is an enzyme—superoxide dismutase—that regulates cellular concentrations of this free radical,2 the importance of free radicals in biology is no longer questionable. These reactive species are common metabolic intermediates that play essential roles in controlling a myriad of physiological functions,3 or are radiolytic reaction products that are cytotoxic.4Identification of free radicals at a cellular level is central to the study of these intermediates. Although there is a wide variety of analytic tools for identifying specific free radicals, spin trapping and EPR spectroscopy have emerged as the primary method to characterize free radicals in real time and at their site of evolution. In 1995 we demonstrated that, in irradiated mouse tumor, HO˙ could be spin trapped and identified using low-frequency EPR spectroscopy in real time and at the site of evolution.5 The spin trapping system consisted of α-(4-pyridyl-1-oxide)-N-tert-butylnitrone [1] in the presence of EtOH (Scheme 1). Hydroxyl radical reacted with EtOH to produce α-hydroxyethyl radical (CH3˙CHOH), which was subsequently spin trapped by nitrone [1] to yield aminoxyl [2].5 Based on a series of in vitro experiments,6 we concluded that only interstitially (i.e. extracellularly) generated HO˙ was spin trapped. In subsequent studies, 5-carboxy-5-methyl-1-pyrroline N-oxide [3] was found to react selectively with HO˙ at near diffusion controlled rates (Scheme 1). Moreover, HO˙ can be spin trapped in aqueous solution, affording nitroxide [4], with as little as 1 Gy of radiation (300 nM HO˙).7
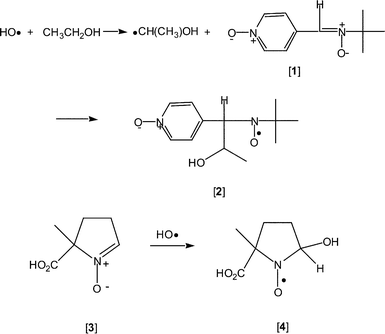 | ||
| Scheme 1 | ||
Effective detection of intracellularly generated HO˙ through spin trapping is feasible only if two conditions can be met. First, the spin trap must be present at high concentration inside the cell, so that the low levels of HO˙ generated intracellularly may be trapped efficiently. Second, the aminoxyls generated by spin trapping must be sufficiently stable in the intracellular environment to allow detection by EPR spectroscopy. Nitrone [3], which specifically traps HO˙, has a pKa of 2.95.8 At physiological pH (≈ 7), nitrone [3] exists essentially entirely as the carboxylate, which cannot diffuse through hydrophobic lipid bilayer membranes and, consequently, cannot enter living cells. This prediction is borne out by the results of the present study, given below. It is possible for carboxylic acids with higher pKas (e.g. fatty acids, pKa ∼5) to permeate biomembranes at physiological pH. We note, however, that when a carboxylate permeates into the cell by passive diffusion, its equilibrium concentration inside the cell relative to the extracellular concentration will be governed by the Nernst equation: Cin/Cout = exp(−zVmF/RT), where z is the ionic charge (−1 for carboxylate), Vm is the membrane potential of the cell, and F, R and T are, respectively, Faraday's constant, the gas constant, and the absolute temperature (RT/F = 26.7 mV at 37 °C). Since Vm for mammalian cells typically range from −60 to −90 mV, we actually expect the intracellular concentration of the carboxylate to be approximately 1/10 to 1/30 of the extracellular concentration; i.e., the carboxylate probe would not accumulate intracellularly to any significant extent by passive permeation alone. Finally, although multiple secondary active transporters exist that move biologically important anions and zwitterions (e.g. amino acids) into cells, none has been identified that transport xenobiotic ions such as spin probes into cells (the well-known transport ATPases of the ABC cassette family actually extrude xenobiotics from cells). In view of the foregoing, the challenge in using nitrone [3] as an intracellular spin trap for HO˙ is: how can we establish a high concentration of nitrone [3] inside a living cell?
We conjecture that nitrone [3] can be converted to its labile acetoxymethyl ester [5], which, being uncharged and hydrophobic, will freely diffuse across the cell membrane to enter the cell (Scheme 2). Subsequent hydrolysis by intracellular esterases9 will yield nitrone [3], as its carboxylate, which, being anionic and hydrophilic, cannot diffuse and escape through the cell membrane, and will be sequestered intracellularly. In order to test this hypothesis, we prepared the aminoxyl acetoxymethyl ester [7] as a model. This enabled us to evaluate hydrolytically labile esters as an effective means to load carboxylate–containing aminoxyls and spin traps into cells, as well as to evaluate the stability of aminoxyls within the intracellular environment.
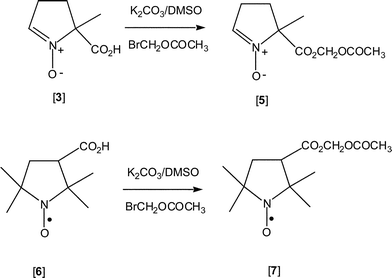 | ||
| Scheme 2 | ||
Results and dscussion
In choosing a hydrolytically labile ester, we decided on an acetoxymethyl (AM) ester, which is readily hydrolyzed to the corresponding carboxylic acid by esterases in most cells, including lymphocytes.9 For these preliminary studies we prepared the AM ester of aminoxyl [6], aminoxyl [7], which has been reported to accumulate in the brains of mice after intravenous administration.10 Modification of the literature procedure10 for the synthesis of aminoxyl [7] resulted in nearly 80% yield of the compound.In the next series of experiments, lymphocytes (1.8 × 107 cells) were incubated with aminoxyl [7] (40 µM), as described in the Experimental section, at ambient temperature for 10–70 min, at which point the cells were centrifuged. Upon removing the loading medium, cells were washed with fresh medium (2 × 10 mL). After the second washing, the cell pellet in each experiment was resuspended in Hanks' Balanced Salt Solution (HBSS; 0.4 mL) and the EPR spectrum of the cell suspension was recorded (an example is Trace A of inset, Fig. 1), as detailed in the Experimental section. The EPR spectrum of the cell suspension was monitored for 30 min and was found to be stable, thus showing no evidence of aminoxyl reduction. Permeabilization of the cells with the non-ionic detergent, digitonin (150 µM, final concentration), in the presence of potassium chromium oxalate (33 mM, final concentration) caused the EPR spectrum to become broadened and nearly undetectable (Trace B of inset, and open triangle, Fig. 1). Lastly, in two control experiments, lymphocytes (1.8 × 107 cells) were incubated with aminoxyl [6] (as the carboxylate) for 50 min, both in the presence and absence of serum, and then washed and resuspended exactly as described above. The EPR spectrum of aminoxyl [6] was undetectable in the cell suspension loaded in the presence of serum (open diamond, Fig. 1) and in the absence of serum (symbol ×, Fig. 1). These findings indicate that the carboxylate of aminoxyl [6] could not readily permeate through the plasma membrane and enter cells. Moreover, this behavior is unaffected by the presence of serum proteins in the loading medium.
![Loading of aminoxyl [7] into Jurkat lymphocytes. Suspensions of Jurkat lymphocytes were loaded with aminoxyl [7] for various amounts of time as described in the Experimental section. The EPR spectral peak height of the middle field peak for each lymphocyte sample after loading is represented by an open circle. Each data point represents the average of one to three measurements of separate suspensions of 1.8 × 107 cells. The solid curve is a least-squares fit of the data to a single exponential of the form y
=
y0
+
Ae−t/τ, with τ
= 1.443t1/2. Two statistics indicate that the data were well-fit by the exponential function: the correlation coefficient was R
= 0.997 and the reduced χ2 value = 0.911. The fit yielded a time to half-maximal loading of t1/2
= 11 ± 2 min; the indicated standard error was obtained through the nonlinear least-squares error matrix. When peak areas obtained by double-integration of the spectra were used instead of peak heights, single-exponential curve-fitting gave essentially the same result (t1/2
= 12 ± 9 min; data not shown for visual clarity). The EPR spectrum of the lymphocyte sample that was loaded for 24 min was taken when the cells were intact (Trace A of inset), and after the cells were permeabilized by 150 µM digitonin in the presence of 33 mM potassium tris(oxalato)chromium(iii) to broaden the spectrum (Trace B of inset); the EPR peak height measured after permeabilization is represented by an open triangle. For the lymphocyte sample that was loaded for 70 min, the EPR spectrum was also recorded on the supernatant cell lysate after cell permeabilization with digitonin, but without the addition of the chromium salt. The EPR peak height is represented by a filled circle. As control, two replicate lymphocyte suspensions were incubated for 50 min with the carboxylate aminoxyl [6], in the presence and absence of serum in the loading medium. The EPR peak height for the sample loaded in the presence of serum is marked by an open diamond; the peak height for the sample loaded in the absence of serum is marked by the symbol ×. All EPR spectra were recorded at room temperature with the following instrumentation settings: microwave power, 20 mW; field set, 3340 G; sweep width, 100 G; modulation frequency, 100 kHz; modulation amplitude, 0.5 G; response time, 0.5 s; sweep, 12.5 G min−1 and receiver gain, 2.5 × 103. The hyperfine splitting constant is AN
= 14.9 G.](/image/article/2004/OB/b310467b/b310467b-f1.gif) | ||
| Fig. 1 Loading of aminoxyl [7] into Jurkat lymphocytes. Suspensions of Jurkat lymphocytes were loaded with aminoxyl [7] for various amounts of time as described in the Experimental section. The EPR spectral peak height of the middle field peak for each lymphocyte sample after loading is represented by an open circle. Each data point represents the average of one to three measurements of separate suspensions of 1.8 × 107 cells. The solid curve is a least-squares fit of the data to a single exponential of the form y = y0 + Ae−t/τ, with τ = 1.443t1/2. Two statistics indicate that the data were well-fit by the exponential function: the correlation coefficient was R = 0.997 and the reduced χ2 value = 0.911. The fit yielded a time to half-maximal loading of t1/2 = 11 ± 2 min; the indicated standard error was obtained through the nonlinear least-squares error matrix. When peak areas obtained by double-integration of the spectra were used instead of peak heights, single-exponential curve-fitting gave essentially the same result (t1/2 = 12 ± 9 min; data not shown for visual clarity). The EPR spectrum of the lymphocyte sample that was loaded for 24 min was taken when the cells were intact (Trace A of inset), and after the cells were permeabilized by 150 µM digitonin in the presence of 33 mM potassium tris(oxalato)chromium(III) to broaden the spectrum (Trace B of inset); the EPR peak height measured after permeabilization is represented by an open triangle. For the lymphocyte sample that was loaded for 70 min, the EPR spectrum was also recorded on the supernatant cell lysate after cell permeabilization with digitonin, but without the addition of the chromium salt. The EPR peak height is represented by a filled circle. As control, two replicate lymphocyte suspensions were incubated for 50 min with the carboxylate aminoxyl [6], in the presence and absence of serum in the loading medium. The EPR peak height for the sample loaded in the presence of serum is marked by an open diamond; the peak height for the sample loaded in the absence of serum is marked by the symbol ×. All EPR spectra were recorded at room temperature with the following instrumentation settings: microwave power, 20 mW; field set, 3340 G; sweep width, 100 G; modulation frequency, 100 kHz; modulation amplitude, 0.5 G; response time, 0.5 s; sweep, 12.5 G min−1 and receiver gain, 2.5 × 103. The hyperfine splitting constant is AN = 14.9 G. | ||
In a final experiment, we used digitonin to lyse the suspension of lymphocytes that had been incubated with aminoxyl AM ester [7] for 70 min and the EPR spectrum of which had been recorded. The suspension was centrifuged to separate lysed cells and debris from the clear supernatant lysate. The EPR spectrum of the supernatant was recorded. As can be seen in Fig. 1, there was ∼37% increase in EPR spectral peak height in the supernatant (filled circle) as compared to intact cells. This is expected, because binding of aminoxyl [6] to organelles and macromolecules, which are present at very high density in intact cells, would retard the motion of spin label and broaden the EPR spectrum, whereas in the very dilute and highly fluid supernatant, the motion of the released aminoxyl is less restricted. By comparing the peak amplitude of the EPR signal from the lysate to a standard calibration curve (details in Experimental section), we estimate that after 70 min of incubation, the average intracellular concentration of aminoxyl [6] was 3.2 mM. Because the cells were incubated with 40 µM of the aminoxyl AM ester [7], this finding suggests that esterase-assisted cleavage enabled the cells to concentrate aminoxyls intracellularly by close to two orders of magnitude.
From the present study, several observations are worth noting. First, aminoxyl [7] could enter the lymphocytes, but the hydrolysis product, aminoxyl [6], could not (Fig. 1, Scheme 3). Second, our data suggest that aminoxyl AM ester [7], once within lymphocytes, was rapidly hydrolyzed to the carboxylate, aminoxyl [6], which accumulated intracellularly. This interpretation is confirmed by the experiment in which lymphocytes incubated for 50 min with the carboxylate aminoxyl [6] failed to accumulate aminoxyls intracellularly. Third, uptake of aminoxyl [7] into these cells was time-dependent. At the shortest incubation time examined (10 min), cellular uptake of aminoxyl [7] was already near half-maximal. Thereafter, steady-state accumulation was reached by about 70 min (Fig. 1). The data were well fit by a single exponential function, with a time to half maximal loading of t1/2 = 11 ± 2 min.11 Fourth, the AM ester approach allows carboxyl-bearing spin probes to accumulate in cells to high concentrations. Finally, once trapped inside lymphocytes, aminoxyl [6] was stable in the intracellular environment. These results indicate that it should be feasible to use the labile ester approach to load spin traps into living cells to detect intracellularly generated HO˙. We are currently synthesizing derivatives of nitrone [3] bearing labile esters.
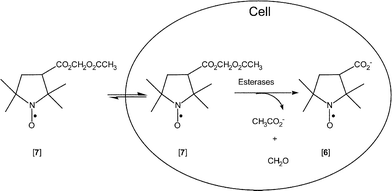 | ||
| Scheme 3 | ||
Experimental
Reagents
3-Carboxy-2,2,5,5-tetramethyl-1-tetramethyl-1-pyrrolidinyloxyl [6] was prepared as described by Rozantsev.12 All reagents were obtained from commercial vendors. EPR spectra were recorded on a Varian Associates, model E-109 spectrometer (Palo Alto, CA). IR spectra were recorded on a FT-IR spectrometer (Perkin-Elmer, Norwalk, CT) in CHCl3. Melting points were obtained on a Thomas Hoover capillary melting point apparatus and were corrected.3-Acetoxymethoxycarbonyl-2,2,5,5-tetramethyl-1-pyrrolidinyloxyl [7]10
To a DMSO (5 mL, dried over CaH2) solution of 3-carboxy-2,2,5,5-tetramethyl-1-tetramethyl-1-pyrrolidinyloxyl [6] (0.5 g, 2.7 mmol) was added anhydrous K2CO3 (0.74 g, 5.4 mmol). This mixture was stirred at room temperature for 5 min at which point bromomethyl acetate (0.45 g, 0.3 mL, 2.94 mmol, Aldrich Chemical, Milwaukee, WI) was added. The reaction was stirred at room temperature for 2 h. A mixture of ice and water was then added and the solution was extracted with CH2Cl2. The organic solution was dried over anhydrous Na2SO4, and evaporated to dryness. Remaining DMSO was removed under high vacuum and the solid that remained in the flask was recrystallized from boiling hexanes to which ether was added dropwise to aid dissolution, yielding aminoxyl [7], mp 72–73 °C (500 mg, 79%). IR (CHCl3): νmax 1764 cm−1.10Loading of lymphocytes with aminoxyls
Replicate suspensions of 1.8 × 107 Jurkat lymphocytes (gift of Dr. Alfredo Garzino Demo) in 10 mL bicarbonate-buffered RPMI 1640 medium (supplemented with 10% v/v fetal bovine serum, 100 units mL−1 penicillin and 100 µg mL−1 streptomycin), were incubated for various periods of time at room temperature with 40 µM aminoxyl [7] and 0.0015% w/v Pluronic F-127 surfactant (BASF Corp., Washington, NJ). After incubation, the cells were centrifuged for 2–3 min at 1000 rpm, and the cell pellet was resuspended and centrifuged twice in 10 mL RPMI 1640 medium, and thereafter resuspended in 400 µL Hanks' Balanced Salt Solution (HBSS) for EPR spectroscopic analysis (i.e. cell density in all experimental samples was 4.25 × 107 cells mL−1). As control, Jurkat lymphocytes were incubated for 50 min at room temperature with 40 µM aminoxyl [6] in RPMI 1640 medium (both with and without fetal bovine serum), and then treated exactly as were the experimental samples. Cell lysis was achieved by adding 1.5 µL of a stock solution of 40 mM digitonin in DMSO to the 400-µL cell suspension. For the 70-min sample, the cell lysate was centrifuged at 15![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 300 rpm for 10 min to sediment cellular debris before EPR spectroscopy. For EPR spectroscopy, samples were added to a quartz flat cell, which was introduced into the cavity of the spectrometer. The quartz cell was open to the atmosphere to allow free equilibration with air.
300 rpm for 10 min to sediment cellular debris before EPR spectroscopy. For EPR spectroscopy, samples were added to a quartz flat cell, which was introduced into the cavity of the spectrometer. The quartz cell was open to the atmosphere to allow free equilibration with air.
Estimation of intracellular concentration of aminoxyl [6] in lymphocytes incubated with AM ester [7]
Jurkat lymphocytes were suspended in HBSS at the same density as was used in all experiments (4.25 × 107 cells mL−1). Digitonin was added to the suspension (150 µM, final concentration) to lyse the cells. The lysate was clarified by centrifugation at 15300 rpm for 10 min to sediment cellular debris. Standard solutions of aminoxyl [6] in this clear cell lysate were prepared by serial dilution to bracket the concentration range of 1–500 µM. An EPR spectrum was acquired for each standard solution; the height of the middle field peak was measured and plotted against the concentration of aminoxyl [6] in the sample to construct a calibration curve. From the EPR spectrum of the clear lysate of the cells that had been incubated with AM ester [7] for 70 min, the middle field peak height was measured and compared with the calibration curve. This comparison showed the concentration of aminoxyl [6] in the 400-µL lysate to be 110 µM. Knowing that the original suspension contained 1.8 × 107 Jurkat cells and that 7.65 × 10−13 L is the average Jurkat cell volume,13 we calculate that the average intracellular concentration of aminoxyl [6] was 3.2 mM. Origin software (OriginLab Corp., Northampton, MA) was used for data analysis and nonlinear least-squares curve fitting.
Acknowledgements
This research was supported in part by grants from the U.S. National Institutes of Health, GM-56481 (J. P. Y. K.) and RR-12257 and EB-2034 (G. M. R.).Notes and references
- (a) R. Gerschman, D. L. Gilbert, S. W. Nye, P. Dwyer and W. O. Fenn, Science, 1954, 119, 623–626 CAS; (b) D. Harman, J. Gerontol., 1956, 11, 298–300 Search PubMed.
- J. M. McCord and I. Fridovich, J. Biol. Chem., 1969, 244, 6049–6055 CAS.
- (a) M. S. Wolin, Arterioscler. Thromb. Vasc. Biol., 2000, 20, 1430–1442 Search PubMed; (b) W. Droge, Physiol. Rev., 2002, 82, 47–95 Search PubMed.
- (a) I. Johnson and P. Howard-Flanders, Radiat. Res., 1965, 24, 184–200; (b) R. Roots and S. Okada, Int. J. Radiat. Biol., 1972, 21, 329–342 CAS; (c) G. Czapski, Isr. J. Chem., 1984, 24, 29–32 CAS.
- H. J. Halpern, C. Yu, E. Barth, M. Peric and G. M. Rosen, Proc. Natl. Acad. Sci. USA, 1995, 92, 796–800 CAS.
- S. Pou, C. L. Ramos, T. Gladwell, E. Renks, M. Centra, D. Young, M. S. Cohen and G. M. Rosen, Anal. Biochem., 1994, 217, 76–83 CrossRef CAS.
- P. Tsai, M. Elas, A. D. Parasca, E. D. Barth, C. Mailer, H. J. Halpern and G. M. Rosen, J. Chem. Soc., Perkin Trans. 2, 2001, 875–880 RSC.
- R. Bonnett, R. F. C. Brown, V. Clark, I. O. Sutherland and A. Todd, J. Chem. Soc., 1959, 2094–2101 RSC.
- R. Y. Tsien, Nature, 1981, 290, 527–528 CrossRef CAS.
- H. Sano, M. Naruse, K.-I. Matsumoto, T. Oi and H. Utsumi, Free Radical Biol. Med., 2000, 28, 959–969 CrossRef CAS.
- This result (t1/2 = 11 ± 2 min) was obtained by fitting the EPR peak height data. We have also double-integrated each EPR spectrum to obtain peak areas. While there was somewhat greater scatter in the peak area data, a single-exponential fit yielded an essentially similar result (t1/2 = 12 ± 9 min).
- E. G. Rozantsev, in Free Nitroxyl Radicals, Plenum Press, New York, 1970, pp. 203–206 Search PubMed.
- C. Miossec-Bartoli, L. Pilatre, P. Peyron, E. N. N'Diaye, V. Collart-Dutilleul, I. Maridonneau-Parini and A. Diu-Hercend, Antimicrob. Agents Chemother., 1999, 43, 2457–2462 CAS.
| This journal is © The Royal Society of Chemistry 2004 |