New isocysteine building blocks and chemoselective peptide ligation
Christian
Dose
and
Oliver
Seitz
*
Institute of Chemistry, Humboldt-University of Berlin, Brook-Taylor-Str. 2, D-12489 Berlin, Germany. E-mail: oliver.seitz@chemie.hu-berlin.de; Fax: +49-30-20937266; Tel: +49-30-20937446
First published on 5th November 2003
Abstract
Boc-, Fmoc- and Cbz-protected isocysteine building blocks were prepared by a concise three-step procedure starting from thiomalic acid. The use of Boc/Trt-protected isocysteine provided convenient access to isocysteinyl peptides that allow the chemoselective ligation of unprotected peptide fragments in water. The pH-dependency of the isocysteine-mediated ligation was compared with that of cysteine-mediated native chemical ligation.
Introduction
The replacement of naturally occurring amino acids by isosteric or artificial amino acids is commonly employed for probing the interaction of a protein with its ligand. Thiol-containing building blocks are of interest due to their particular reactivity which mediates inhibitory interactions with protein metallo centers, conformational restriction through disulfide bond formation and site-selective tagging reactions among other effects. The β-mercapto amino acid cysteine is by far the most frequently used thiol-containing building block. Its 1,2-aminothiol structure also makes it an invaluable tool for the efficient and convenient joining of two unprotected peptide fragments.1–5The introduction of α-mercapto acids provides the opportunity to alter thiol acidity and/or thiolate basicity which can allow the fine-tuning of metal–ligand interactions. For example, some inhibitors of metalloproteases such as angiotensin-converting enzymes contain α-mercapto acyl peptides.6,7 The β-amino-α-mercapto amino acid isocysteine is a useful cysteine analogue and has been shown to increase the inhibitory activity of peptidic stromelysin binders when incorporated as a replacement of cysteine.8 Isocysteine also might serve as a replacement of β3-H cysteine in β-peptides which opens the possibility of altering thiol reactivity without changing the backbone length.9 There is, however, only one report that describes the preparation of an isocysteine derivative that suits the needs of solid-phase peptide synthesis.8 We here present convenient syntheses of Boc-, Fmoc- and Cbz-protected isocysteine building blocks. Furthermore we show the utility of isocysteine to support the ligation of unprotected peptide segments by a native chemical ligation-like reaction. To illustrate the effects of alterations of the thiol acidity the pH-dependency of the isocysteine ligation was compared with that of cysteine-mediated native chemical ligation.
Results and discussion
Hanglows’ synthesis of isocysteine 7 and also our route to N,S-protected building blocks 8–11 was based on commercially available DL-thiomalic acid 1 as described by Gustavson.8,10,11 According to this, simultaneous protection of both the α-mercapto and the α-carboxylic group was achieved upon reaction with cyclopentanone in the presence of p-toluenesulfonic acid (Scheme 1). The β-carboxyl group in the formed oxathiolanone 2 remained unaffected and was subsequently converted to the acyl azide. In contrast to Hanglows’ Fmoc/Trt-Isocys synthesis, in which the isocyanate 3 formed after the Curtius rearrangement was hydrolysed to obtain isocysteine in unprotected form, it was preferred to fashion a more direct establishing of Fmoc-, Cbz- and Boc-protecting groups. The isocyanate 3 was allowed to react with 9-fluorenylmethanol, benzyl alcohol or tert-butanol, which completed a three-step sequence and furnished the fully protected isocysteine derivatives 4, 5 and 6 in 77–87% yield. We next sought a method that would liberate both the carboxyl group and the α-mercapto group without detriment to the acid- and base-sensitive protecting groups in 4–6. The opening of the cyclic ketal was initiated by ester hydrolysis with aqueous LiOH in THF, which proved straightforward with Cbz- and Boc-protected isocysteines 4 and 6. The presence of the base-sensitive Fmoc-group in 5 rendered this step challenging. It was found that careful addition of exactly one equivalent of lithium hydroxide in THF, however, enabled a clean and quantitative removal of the ketal group. The liberated α-mercapto groups were subsequently masked by omitting any attempts to isolate the N-protected isocysteine derivatives. Instead, it was preferred to directly add p-methoxybenzyl chloride or trityl bromide to the basic THF solution obtained after ester hydrolysis. This convenient one-pot procedure afforded the known Fmoc/Trt-protected isocysteine 7 and the novel Fmoc/PMB-, Cbz/PMB-, Boc/Trt- and Boc/PMB-protected building blocks 8–11 in 50–96% yield. It is noted that the same strategy would allow for the synthesis of enantiomerically pure isocysteine if D- or L-thiomalic acid were used.12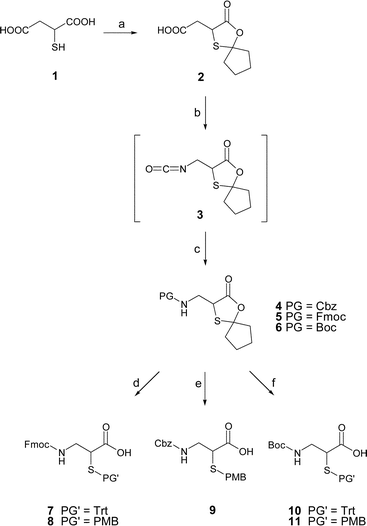 | ||
| Scheme 1 a) cyclopentanone, p-TsOH, PhCH3, reflux, 68%; b) 1. diphenylphoshoryl azide, TEA, PhCH3; 2. 85 °C; c) PhCH2OH, 85 °C, 86% (3→4); 9-fluorenylmethanol, 85 °C, 87% (3→5); tBuOH, 75 °C, 77% (3→6); d) 1. 0.2 M LiOH, THF, 0 °C; 2. Trt-Br, 50% (5→7); 1. 0.2 M LiOH, THF, 0 °C; 2. PMB-Cl, 80% (5→8); e) 1. 1 M LiOH, THF, RT; 2. PMB-Cl, 96%; f) 1. 1 M LiOH, THF; 2. Trt-Br, 78% (6→10); 1. 1 M LiOH, THF, RT. 2. PMB-Cl, 77% (6→11). | ||
Having established expedient access to various protected isocysteine derivatives we next set out to explore their utility in the solid-phase synthesis of isocysteine containing peptides. Previous work had demonstrated the usefulness of the Fmoc/Trt-protected building block 7.8 We envisaged investigating the use of isocysteine as a ligation handle (vide infra). Our particular interest, therefore, concerned the synthesis of peptides such as 16 bearing isocysteine as N-terminal amino acid. We reckoned that Cbz/PMB- and Boc/PMB-protected isocysteine building blocks 9 and 11 would enable the isocysteinyl-peptide 16 to be assembled by applying the Boc-strategy. For the final cleavage trifluoromethanesulfonic acid was employed as a non-toxic alternative to HF. The crude peptide obtained after cleavage, however, failed to precipitate upon addition of ether which hampered its purification. After repeated unsuccessful attempts with various scavengers, we chose to couple Boc/Trt-protected isocysteine 10 to resin-bound peptide 14 which was assembled by using Fmoc/tBu-protected amino acids (Scheme 2). It was expected that the application of the Fmoc-strategy and TFA as cleaving agent would be beneficial. TFA in contrast to TFMSA (trifluoromethanesulfonic acid) is volatile and allows vacuum concentration thereby facilitating ether precipitation. Indeed, in this case purification was straightforward providing peptide 16 after HPLC purification in 69% overall yield.
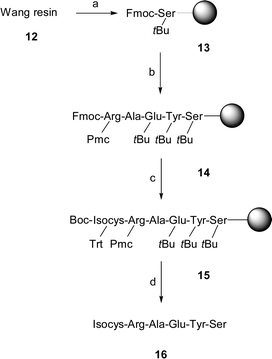 | ||
| Scheme 2 a) 10 eq. Fmoc-Ser(tBu)-OH, 5 eq. DIC, 0.1 eq. DMAP, DMF; b) 1. DMF/piperidine (4 : 1), 2. 4 eq. Fmoc-Aa-OH, 4 eq. HOBt, 3.6 eq. HBTU, 8 eq. DIPEA; 3. pyridine/Ac2O (5 : 1); c) 1. DMF/piperidine (4 : 1), 2. 4 eq. Boc-Isocys(Trt)-OH, 4 eq. HOBt, 3.6 eq. HBTU, 8 eq. DIPEA; 3. pyridine/Ac2O (5 : 1); d) CF3COOH/PhSMe/Et3SiH/H2O (92 : 3 : 3 : 2), 69%. | ||
Terminally modified peptides such as bromoacetamido-,13 maleinimido-,14 hydroxylamino-15 and azidopeptides16,17 among others18 are invaluable tools that can allow chemoselective ligation of two unprotected peptide fragments to be performed in aqueous solution. The 1,2-aminothiol structure of N-terminal cysteine is one of the most suitable entities for achieving such segment couplings demonstrated best by the numerous protein syntheses that have been enabled by the native chemical ligation technology.2,19 This reaction (Scheme 3) is initiated by a reversible thiol-exchange reaction of cysteine conjugate 17 with thioester 18.20 The formed thioester intermediate 19 is subject to a spontaneous S–N-acyl shift, which establishes the α-peptide bond in product 20. In isocysteine, the 1,2-aminothiol structure is retained. Accordingly, isocysteine is expected to support fragment ligations by a native chemical ligation-like reaction mechanism.21 The S–N-acyl shift in thioester-intermediate 22 would lead to the formation of a β-peptide bond in 23. The isocysteine-mediated native chemical ligation would hence offer prospects for a convergent assembly of peptides with β-amino acid architecture.9,22,23
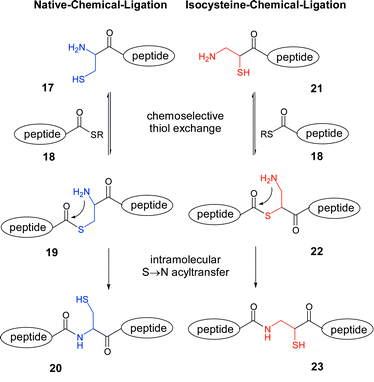 | ||
| Scheme 3 | ||
To assess the feasibility of isocysteine-mediated native chemical ligation we explored model peptides and ligation conditions as described by Kent and co-workers.3,24 First, peptide mercaptopropionamide thioester 24, which was obtained through solid-phase synthesis on a mercaptopropionic acid loaded Rink-resin, was converted to the more reactive mercaptobenzylester 25. Peptide thioester 25 was allowed to react with the isocysteine-peptide 16. Reactions were carried out in argon saturated 0.1 M phosphate buffer and in the presence of 4% benzylmercaptan which helped to prevent thioester hydrolysis and to maintain a reducing environment. At pH 7 and 1 mM concentration of reactants the isocysteine-mediated native chemical ligation proceeded smoothly. HPLC/ESI-MS analysis showed a new peak that appeared at 16 min with an m/z that corresponded to ligation product 26 (Fig. 1). The ligation product formed lacked susceptibility to attempted hydrolysis by NaOH-treatment which provided support for the formation of a non-hydrolysable peptide bond. It can be concluded that isocysteine supports a native chemical ligation-like reaction. It is interesting to note that the S→N acyl transfer likely to be involved in the isocysteine ligation proceeds via main chain expansion as opposed to S→N acyl transfer involved in cysteine and auxiliary mediated ligations25–29 that are characterised by main chain contraction. This feature might be of less importance in convergent peptide assembly, but we presume that it could affect ligation reactions that proceed under geometrical constraints such as peptide-30,31 and DNA-template controlled reactions.32,33
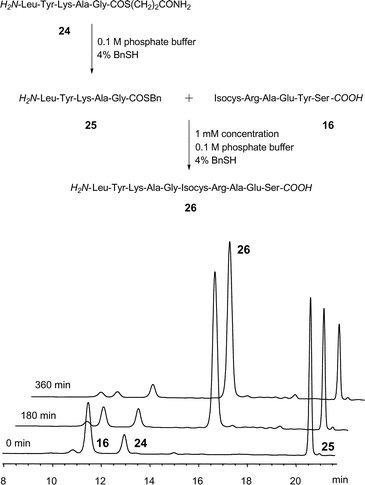 | ||
| Fig. 1 Analytical HPLC trace of the ligation reaction at λ = 280 nm (16 + 25) and pH 7. | ||
It is worthwhile noting that both the isocysteine containing starting material 16 and the ligation product 26 exist as a mixture of diastereomers due to the use of racemic isocysteine. Nevertheless, under the selected conditions the HPLC-traces showed single albeit broadened peaks and it was therefore not possible to explore whether one of the diastereomers conferred higher ligation rates than the other.
Fig. 2 shows the time-dependent formation of ligation product 26 at pH 7 and pH 6. It became evident that the ligation at pH 6 is slower than the ligation at pH 7. For example, product 26 was formed in 63% yield after 240 min as opposed to 44% yield obtained at pH 6.
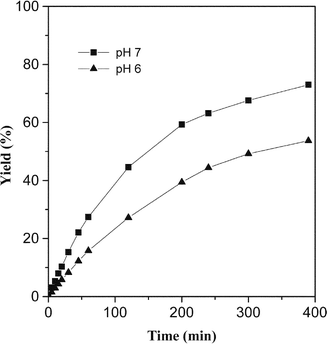 | ||
| Fig. 2 Isocysteine-mediated chemical ligation at pH 7 and pH 6. | ||
It has been shown that the cysteine-mediated native chemical ligation proceeds rapidly around pH 7 but is rendered less efficient at pH < 5.5.34 To explore whether the isocysteine-mediated native chemical ligation would exhibit a similar pH-dependence we compared the initial rate of formation of isocysteine-peptide 26 and cysteine-containing peptide 28 as a function of pH at 1 mM concentrations of reactants and at identical reaction conditions (Fig. 3). RP-HPLC-analysis revealed that the initial rate of cysteine native chemical ligation was reduced by a factor of 0.25 when lowering the pH from pH 7 to pH 6. This pH decrease reduced the initial rate of isocysteine native chemical ligation only by a factor of 0.56. Further pH decreases to pH 5 led to a diminution of initial rates such that the formation of ligation products almost ceased. At pH 7, the ligation of cysteine-peptide 28 was 3-times faster than ligation of the isocysteine-peptide 26 while at pH < 6, the isocysteine native chemical ligation was as efficient as the cysteine native chemical ligation.
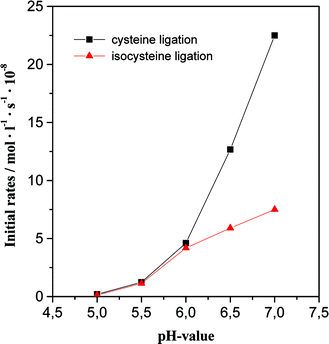 | ||
| Fig. 3 pH-Profile of initial ligation rates of the cysteine ligation (25 + Cys-Arg-Ala-Glu-Tyr-Ser, 27 → Leu-Tyr-Lys-Ala-Gly-Cys-Arg-Ala-Glu-Tyr-Ser, 28) and the isocysteine ligation (16 + 25 → 26). Ligation conditions as specified in Fig. 1. | ||
It became apparent that the isocysteine-mediated ligation is less pH-dependent than the cysteine-mediated native chemical ligation. We assume that the increased acidity of the thiol group is the decisive factor that leads to this attenuation of the pH-dependence. This assumption is based on the notion that native chemical ligation proceeds through the thiolate form of the cysteine-peptide and that the thiol-exchange reaction is rate-limiting.3 In isocysteine, the α-thiol group should be more acidic than the β-thiol group of cysteine due to the inductive effect of the carboxyl group. This influence becomes manifest in the pKa-value of thioglycolic amide (pKa = 8.2) which is considerably lower than that of β-mercaptopropionic amide (pKa = 9.4).35 The calculated pKa-values of isocysteine amide (pKa = 8.2) and cysteine amide (pKa = 9.1) suggest a similar difference of thiol acidity.36 The more acidic the thiol group, the more thiolate is provided even under acidic conditions and hence the faster the ligation reaction. On the other hand, inductive and steric effects are expected to decrease the nucleophilicity of the α-thiolate in isocysteine when compared with the β-thiolate of cysteine which might explain why at pH > 6 the cysteine native chemical ligation proved faster than the isocysteine native chemical ligation.
Conclusion
In conclusion, we have provided convenient synthetic access to various isocysteine building blocks for solution and solid-phase peptide synthesis. The replacement of cysteine by isocysteine allows for an alteration of thiol reactivity as evidenced by the attenuated pH-dependence of isocysteine-mediated native chemical ligation of unprotected peptide fragments. Isocysteine being a β-amino acid, also might be incorporated in β-peptides, structurally well-defined peptide analogues that show interesting biological activities. It is, for example, conceivable that the replacement of β3-H-cysteine by isocysteine allows the thiol reactivity to be altered while maintaining the backbone length and the ability to confer chemoselective fragment condensations. In this regard, the ability of isocysteine to support native chemical ligation reactions might be of advantage for the convergent assembly of modified β-peptides.Experimental
General materials and methods
Reagents and chemicals were obtained from Acros, Aldrich, Biosolve, Fluka, Novabiochem or Senn Chemicals and were used without further purification. All solvents were freshly distilled. Proton and carbon NMR spectra were recorded on a Varian Mercury 400 or a Bruker DRX-500 spectrometer at room temperature. The signals of the residual protonated solvent (CDCl3 or [D6]DMSO) were used as reference signals. Coupling constants J are reported in Hz. Flash chromatography was performed using Merck silica gel 60. TLC was performed with aluminium-backed silica gel 60 F254 plates (Merck). Melting points were measured using a Büchi B-540 apparatus in open capillary tubes and are not corrected.Reversed phase HPLC
Analytical reversed phase HPLC was performed with an Agilent 1100 series system and a DAD-detector recording at 280 nm. A Macherey & Nagel Nucleosil (100-5) type CC 250/4 Nautilus RP-18 column was used at room temperature. Semi-preparative reversed phase HPLC was performed with a Gilson Nebula Series with automated fraction collection and Gilson 156 UV-detector. A Macherey & Nagel SP 125/10 Nucleodur Gravity RP-18 column was used at 55 °C. Gradients of solvent A (98.9% water/1% acetonitrile/0.1% TFA) and B (98.9% acetonitrile/1% water/0.1% TFA) were adjusted according to the separation needs.Mass spectrometry
Matrix assisted laser desorption ionisation analysis was carried out with a Voyager-DE Pro BioSpectrometer™ from PerSeptive Biosystems using a 2,5-dihydroxybenzoic acid (DHB) matrix. ESI-MS was carried out using a Agilent 1100 series system with a Macherey & Nagel Nucleosil (100-5) type CC 250/4 Nautilus RP-18 column and a Finnigan Thermoquest LCQ. FAB-MS was recorded on a JEOL JMS-SX102A machine, applied matrices were given for each compound.Solid-phase peptide synthesis
Reactions were carried out at room temperature and performed manually using 5 ml polyethylene syringe reactors which were equipped with a fritted disc. All peptides were synthesised using HBTU/HOBt chemistry. Typically 4 eq. of the corresponding amino acid were allowed to react for 2–3 min with 3.6 eq. of HBTU, 4 eq. of HOBt and 8 eq. of DIPEA in DMF/CH2Cl2 (1 : 1). The solution was added to the resin and agitated for 2 h at room temperature. Cysteine was coupled using DIC/HOBt chemistry in order to avoid extensive racemisation. Typically 4 eq. of the amino acid were reacted with 4 eq. of HOBt and 4 eq. of DIC in DMF for 20 min and then added to the resin. Unreacted amino groups were capped by a 15 min treatment of pyridine/Ac2O (5 : 1) after every coupling step. Fmoc cleavage was achieved using two times a solution of 20% piperidine in DMF for 10 min. The Boc group was cleaved using two times pure TFA for 5 min. The resins were washed after every step with CH2Cl2 (5×), DMF (5×) and CH2Cl2 (5×) to avoid any side reactions.(2-Oxo-1-oxa-4-thia-spiro[4.4]non-3-yl)-acetic acid (2)
To a solution of 2-mercaptosuccinic acid 1 (5.0 g, 33.3 mmol) in toluene (70 ml) were added cyclopentanone (3.54 ml, 40.2 mmol) and p-toluenesulfonic acid (76 mg, 0.40 mmol). The reaction vessel was equipped with a Dean–Stark trap and was refluxed for 8 h. The solution was concentrated under reduced pressure, and the residue was dissolved in sat. NaHCO3 (30 ml). The aqueous solution was washed with CH2Cl2 (2 × 20 ml), adjusted to pH 2 with concentrated HCl and extracted with CH2Cl2 (3 × 30 ml). The organic phase was dried (MgSO4) and evaporated under reduced pressure. The residue was purified by silica gel flash chromatography to furnish 4.90 g (68%) of 2 as a white solid. Mp = 93 °C. Rf = 0.21 (EtOAc : cyclohexane) = 2 : 5 + 1% AcOH). 1H-NMR (CDCl3): δ 1.75 (m, 4H); 1.94 (m, 2H); 2.24 (m, 2H); 2.84 (dd, 1H, J = 9.16, J = 17.69); 3.21 (dd, 1H, J = 4.02, J = 17.57); 4.38 (dd, 1H, J = 4.14, J = 9.16); 11.43 (s, 1H). 13C-NMR (CDCl3): 24.89 (CH2); 25.17 (CH2); 39.15 (CH2); 42.63 (CH2); 43.94 (CH2); 44.93 (CH); 97.20 (Cq); 174.83 (Cq); 177.59 (Cq). HRMS [FAB, m-NBA, pos.]: calcd. 216.0456; found 216.0468.(2-Oxo-1-oxa-4-thia-spiro[4.4]non-3-ylmethyl)-carbamic acid benzyl ester (4)
A solution of 2 (1.02 g, 4.73 mmol) and triethylamine (728 µl, 5.20 mmol) in toluene (20 ml) was stirred 30 min at room temperature. The reaction mixture was cooled to 0 °C and diphenylphosphoryl azide (1.12 ml, 5.10 mmol) was added dropwise. After stirring for 2 h at room temperature, the solution was heated to 85 °C until the evolution of N2 ceased. The reaction mixture was cooled to room temperature and EtOAc (30 ml) was added. The organic phase was washed with sat. NaHCO3 (20 ml), dist. H2O (20 ml), dried (MgSO4) and concentrated under reduced pressure. The residue was dissolved in toluene (20 ml) and benzyl alcohol (460 µl, 4.45 mmol) was added. The solution was kept at 80 °C for 14 h and then allowed to cool to room temperature. The solvent was evaporated under reduced pressure and the residue was purified by silica gel chromatography to afford 1.32 g (87%) of 4 as an yellow oil. Rf = 0.24 (EtOAc : cyclohexane = 4 : 1). 1H-NMR (CDCl3): δ 1.75 (m, 4H); 1.94 (m, 2H); 2.24 (m, 2H); 3.65 (m, 2H); 4.19 (dd, 1H, J = 5.65); 5.07 (s, 2H); 7.31 (m, 5H). 13C-NMR (CDCl3): 23.58 (CH2); 23.84 (CH2); 41.34 (CH2); 42.53 (2 × CH2); 48.29 (CH2); 67.12 (CH2); 95.85 (Cq); 128.24–128.73 (4 × CH); 136.33 (Cq); 156.42 (Cq); 173.43 (Cq). HRMS [FAB, m-NBA, pos.]: calcd. 321.1035; found 321.1020.(2-Oxo-1-oxa-4-thia-spiro[4.4]non-3-ylmethyl)-carbamic acid 9H-fluoren-9-ylmethyl ester (5)
A solution of 2 (200 mg, 0.92 mmol) and triethylamine (142 µl, 1.01 mmol) in toluene (10 ml) was stirred 30 min at room temperature. The reaction was cooled to 0 °C and diphenylphosphoryl azide (198 µl, 0.92 mmol) was added dropwise. After stirring over 2 h at room temperature, the solution was heated up to 85 °C until the evolution of N2 ceased. The reaction was cooled to room temperature and EtOAc (20 ml) was added. The organic phase was washed with sat. NaHCO3 (15 ml), dist. H2O (15 ml), dried (MgSO4) and concentrated under reduced pressure. The dried residue was dissolved in toluene (10 ml) and 9-fluorenylmethanol (157 mg, 0.8 mmol) was added. The solution was kept at 80 °C for 24 h and then allowed to cool to room temperature. The solvent was evaporated under reduced pressure and the residue was purified by silica gel chromatography to afford 320 mg (85%) of 5 as an yellow oil. Rf = 0.22 (EtOAc : cyclohexane = 4 : 1). 1H-NMR (CDCl3): δ 1.75 (m, 4H); 1.94 (m, 2H); 2.24 (m, 2H); 3.66 (m, 2H); 4.20 (m, 1H); 4.37 (m, 2H); 7.28 (t, 2H, J = 7.40); 7.37 (t, 2H, J = 7.53); 7.56 (d, 2H, J = 7.53); 7.73 (d, 2H, J = 7.53). 13C-NMR (CDCl3): 23.67 (CH2); 23.95 (CH2); 41.42 (CH2); 42.53 (CH2); 42.66 (CH); 47.32 (CH); 48.36 (CH2); 67.28 (CH2); 95.70 (Cq); 120.22–127.93 (8 × CH); 130.01 (2 × Cq); 141.50 (Cq); 143.95 (Cq); 156.51 (Cq); 173.62 (Cq). HRMS [FAB, m-NBA, pos.]: calcd. 409.1348; found 409.1335.(2-Oxo-1-oxa-4-thia-spiro[4.4]non-3-ylmethyl)-carbamic acid tert-butyl ester (6)
A solution of 2 (200 mg, 0.70 mmol) and triethylamine (108 µl, 0.77 mmol) in toluene (10 ml) was stirred 30 min at room temperature. The reaction was cooled to 0 °C and diphenylphosphoryl azide (160 µl, 0.77 mmol) was added dropwise. After stirring over 2 h at room temperature, the solution was heated to 85 °C until the evolution of N2 ceased. The reaction was cooled to room temperature and EtOAc (20 ml) was added. The organic phase was washed with sat. NaHCO3 (15 ml), dist. H2O (15 ml), dried (MgSO4) and concentrated under reduced pressure. The residue was dissolved in toluene (10 ml) and tert-butanol (881 µl, 7.7 mmol) was added. The solution was kept at 75 °C for 40 h and then allowed to cool to room temperature. The solvent was evaporated under reduced pressure and the residue was purified by silica gel chromatography to afford 183 mg (73%) of 6 as an yellow oil. Rf = 0.26 (EtOAc : cyclohexane = 4 : 1). 1H-NMR (CDCl3): δ 1.42 (s 9H); 1.75 (m 2H); 1.94 (m, 2H); 2.24 (m, 2H); 3.59 (m, 2H); 4.18 (m, 1H). 13C-NMR (CDCl3): 23.57 (CH2); 23.83 (CH2); 28.42 (CH3); 41.44 (CH2); 42.19 (CH2); 42.53 (2 × CH2); 48.35 (CH); 80.02 (Cq); 95.79 (Cq); 173.54 (Cq). HRMS [FAB, m-NBA, pos.]: calcd. 287.1191; found 287.1186.3-(9H-Fluoren-9-ylmethoxycarbonylamino)-2-tritylsulfanyl-propionic acid (7)
To a solution of 5 (80 mg, 0.20 mmol) in THF (5 ml), cooled to 0 °C was added dropwise 0.2 M LiOH (1 ml). After 30 min the starting material was completely consumed and trityl bromide (110 mg, 0.40 mmol) was added. The solution was stirred 3 h at room temperature and adjusted to pH ∼ 3 with AcOH. The aqueous solution was extracted with EtOAc (3 × 10 ml). The organic extract was dried (MgSO4), filtered and concentrated under reduced pressure. The residue was purified by silica gel flash chromatography to furnish 57 mg (50%) of 7 as a white solid. Mp = 128–130 °C. Rf = 0.36 (DCM : MeOH = 95 : 5 + 1% AcOH). 1H-NMR ([D6]-DMSO): δ 2.99 (m, 1H); 3.05 (m, 2H); 4.11–4.18 (m, 3H); 7.30–7.37 (m, 19H); 7.64 (t, 2H, J = 8.48); 7.87 (d, 2H, J = 7.48). 13C-NMR ([D6]-DMSO): 36.54 (CH); 46.55 (CH); 67.40 (Cq); 79.80 (CH2); 126.89–129.24 (6 × CH); 140.68 (Cq); 143.79 (Cq); 144.17 (Cq); 155.60 (Cq); 176.94 (Cq). HRMS [FAB, m-NBA, pos.]: calcd. 585.1974; found 585.1978.3-(9H-Fluoren-9-ylmethoxycarbonylamino)-2-(4-methoxy-benzylsulfanyl)-propionic acid (8)
To a solution of 5 (80 mg, 0.20 mmol) in THF (5 ml), cooled to 0 °C was added dropwise 0.2 M LiOH (1 ml). After 30 min the starting material was completely consumed and p-methoxybenzyl chloride (28 µl, 0.20 mmol) was added. The solution was stirred 2 h at 0 °C and adjusted to pH ∼ 3 with AcOH. The aqueous solution was extracted with EtOAc (3 × 10 ml). The organic extract was dried (MgSO4), filtered and concentrated under reduced pressure. The residue was purified by silica gel flash chromatography to furnish 73 mg (80%) of 8 as a yellow oil. Rf = 0.35 (DCM : MeOH = 95 : 5 + 1% AcOH). 1H-NMR ([D6]-DMSO): δ 3.24 (m, 1H); 3.34 (m, 2H); 3.69 (m, 3H); 3.77 (m, 2H); 4.24 (m, 3H); 6.82 (d, 2H, J = 8.53); 7.23 (d, 2H, J = 8.53); 7.31 (t, 2H, J = 7.15); 7.40 (t, 2H, J = 7.40); 7.69 (t, 2H, J = 6.40); 7.88 (d, 2H, J = 7.53). 13C-NMR ([D6]-DMSO): 34.31 (CH2); 41.98 (CH); 46.25 (CH2); 46.98 (CH); 55.30 (CH3); 65.92 (CH2); 114.12 (2 × CH); 120.46 (2 × CH); 125.58–129.73 (8 × CH); 130.46 (Cq); 141.05 (2 × Cq); 144.17 (2 × Cq); 156.48 (Cq); 158.59 (Cq); 172.73 (Cq). HRMS [FAB, m-NBA, pos.]: calcd. 463.1453; found 463.1432.3-Benzyloxycarbonylamino-2-(4-methoxy-benzylsulfanyl)-propionic acid (9)
To a solution of 4 (1.29 g, 4.01 mmol) in 20 ml THF was added at 0 °C dropwise 1 M LiOH (15 ml) and the reaction was stirred for 1 h at room temperature. The mixture was treated with p-methoxybenzyl chloride (544 µl, 4.01 mmol) and stirred at room temperature for further 3 h. The reaction was adjusted to pH ∼ 3 with AcOH and H2O (10 ml) was added. The aqueous solution was extracted with EtOAc (3 × 20 ml). The organic phase was dried (MgSO4) and evaporated under reduced pressure. The residue was purified by silica gel flash chromatography to obtain 1.45 g (96%) of 9 as a yellow oil. Rf = 0.34 (DCM : MeOH = 95 : 5 + 1% AcOH). 1H-NMR ([D6]-DMSO): δ 3.38 (m, 2H); 3.47 (m, 2H); 3.75 (s, 3H); 3.83 (m, 2H); 5.05 (s, 2H); 6.81 (d, 2H, J = 8.53); 7.23 (d, 2H, J = 8.53); 7.32 (m, 5H). 13C-NMR ([D6]-DMSO): 35.40 (CH2); 41.04 (CH2); 44.99 (CH); 55.15 (CH3); 66.94 (CH2); 113.95 (2 × CH); 128.04–128.45 (4 × CH); 130.18 (2 × CH); 136.07 (Cq); 156.24 (Cq); 158.82 (Cq); 176.28 (Cq). HRMS [FAB, m-NBA, pos.]: calcd. 375.1140; found 375.1160.3-tert-Butoxycarbonylamino-2-tritylsulfanyl-propionic acid (10)
To a solution of 6 (344 mg, 1.17 mmol) in 10 ml THF was added dropwise 1 M LiOH (5 ml) at 0 °C. The mixture was heated to room temperature, stirred for 1 h before trityl bromide (388 mg, 1.2 mmol) was added. The solution was stirred for further 4 h and adjusted to pH ∼ 3 by AcOH. H2O (10 ml) was added and the aqueous solution was extracted with EtOAc (3 × 10 ml). The organic phase was dried (MgSO4), concentrated under reduced pressure and the residue was purified by silica gel flash chromatography to furnish 419 mg (78%) of 10 as a white solid. Mp = 58–60 °C. Rf = 0.32 (DCM : MeOH = 95 : 5 + 1% AcOH). 1H-NMR ([D6]-DMSO): δ 1.46 (s, 9H); 2.99 (m, 2H); 3.07 (m, 1H); 7.25–7.36 (m, 15H). 13C-NMR ([D6]-DMSO): 28.17 (3 × CH3); 36.43 (CH2); 47.07 (CH); 67.35 (Cq); 77.69 (Cq); 126.93 (3 × CH); 128.08 (3 × CH); 129.21 (3 × CH); 144.15 (Cq); 155.01 (Cq); 172.28 (Cq). HRMS [FAB, m-NBA, pos.]: calcd. 463.1817; found 463.1835.3-tert-Butoxycarbonylamino-2-(4-methoxy-benzylsulfanyl)-propionic acid (11)
To a solution of 6 (90 mg, 0.31 mmol) in 5 ml THF was added dropwise 1 M LiOH (3 ml) at 0 °C. The mixture was heated to room temperature, stirred for 1 h before p-methoxybenzyl chloride (43 µl, 0.31 mmol) was added. The solution was stirred for further 3 h and adjusted to pH ∼ 3 by AcOH. H2O (10 ml) was added and the aqueous solution was extracted with EtOAc (3 × 10 ml). The organic phase was dried (MgSO4), concentrated under reduced pressure and the residue was purified by silica gel flash chromatography to furnish 81 mg (77%) of 11 as a yellow oil. Rf = 0.34 (DCM : MeOH = 95 : 5 + 1% AcOH). 1H-NMR ([D6]-DMSO): δ 1.36 (s, 9H); 3.18 (m, 2H); 3.28 (m, 2H); 3.72 (s, 3H); 3.75 (m, 2H); 6.86 (d, 2H); 7.23 (d, 2H). 13C-NMR ([D6]-DMSO): 28.24 (3 × CH3); 33.92 (CH2); 46.13 (CH2); 55.05 (CH3); 62.61 (CH); 77.69 (Cq); 77.95 (Cq); 113.46 (CH); 113.83 (CH); 127.97 (Cq); 129.46 (CH); 130.18 (CH); 155.59 (Cq); 158.34 (Cq); 172.44 (Cq). HRMS [FAB, m-NBA, pos.]: calcd. 341.1297; found 341.1282.Isocys-Arg-Ala-Glu-Tyr-Ser (16)
52 mg of Wang resin (0.98 mmol g−1, 0.051 mmol) was used and the peptide assembly proceeded as described above (Fmoc-Ser(t-Bu)-OH, Fmoc-Tyr(t-Bu)-OH, Fmoc-Glu(t-Bu)-OH, Fmoc-Ala-OH, Fmoc-Arg(Pmc)-OH, Boc-Isocys(Trt)-OH). The peptide was released and deprotected by treating the resin with a solution of CF3COOH : PhSMe : Et3SiH : H2O (92 : 3 : 3 : 2, 2 ml) for 2 h. After filtration the resin was washed two times with pure TFA (1 ml). The combined filtrates were concentrated under reduced pressure and cold ether was added (5 ml). The precipitate was collected by centrifugation and washed two times with ether. Purification by semi-preparative reversed phase HPLC and lyophilization furnished 33 mg (69%) of 16 as a white powder. MS (MALDI-TOF, DHB, pos.): m/z: calcd. for C29H46N9O11S [M + H]: 728.8, found: 729.1.Leu-Tyr-Lys-Ala-Gly-COS(CH2)2CONH2 (24)
To 104 mg of MBHA resin (0.8 mmol g−1, 0.083 mmol) was added a solution of 3-tritylsulfanylpropionic acid (1.47 mg, 0.42 mmol), HBTU (159.3 mg, 0.42 mmol) and DIPEA (145 µl, 0.83 mmol) in DMF/DCM (1 : 1, 2 ml). After 14 h the trityl group was removed by means of a one hour treatment with CF3COOH : Et3SiH (95 : 5, 1 ml). The peptide assembly proceeded as described above (Boc-Gly-OH, Boc-Ala-OH, Boc-Lys(2-Cl-Z)-OH, Boc-Tyr(Bz)-OH, Boc-Leu-OH). The peptide was released and deprotected by treating the resin with a solution of CF3COOH : CF3SO3H : PhSMe : EDT (77 : 10 : 10 : 3, 2 ml) for 2 h. After filtration the resin was washed two times with pure TFA (1 ml). The combined filtrates were concentrated under reduced pressure and cold ether was added (5 ml). The precipitate was collected by centrifugation and washed two times with ether. Purification by semi-preparative reversed phase HPLC and lyophilization furnished 35 mg (49%) of 24 as a white powder. MS (MALDI-TOF, DHB, pos.): calcd. for C29H48N7O7S [M + H]: 638.8 found: 639.6.Cys-Arg-Ala-Glu-Tyr-Ser (27)
52 mg of Wang resin (0.98 mmol g−1, 0.051 mmol) were used and the peptide assembly proceeded as described above (Fmoc-Ser(t-Bu)-OH, Fmoc-Tyr(t-Bu)-OH, Fmoc-Glu(t-Bu)-OH, Fmoc-Ala-OH, Fmoc-Arg(Pmc)-OH, Fmoc-Cys(Trt)-OH). The peptide was released and deprotected by treating the resin with a solution of CF3COOH : PhSMe : Et3SiH : H2O (92 : 3 : 3 : 2, 2 ml) for 2 h. After filtration the resin was washed two times with pure TFA (1 ml). The combined filtrates were concentrated under reduced pressure and cold ether was added (5 ml). The precipitate was collected by centrifugation and washed two times with ether. Purification by semi-preparative reversed phase HPLC and lyophilization furnished 38 mg (78%) of 27 as a white powder. MS (MALDI-TOF, DHB, pos.): m/z: calcd. for C29H46N9O11S [M + H]: 728.8, found: 729.2.Chemical ligations
Leu-Tyr-Lys-Ala-Gly-COS(CH2)2CONH2 (24) (100 µl of 10 mM solution in H2O) was added to an argon saturated 0.1 M sodium phosphate buffer (800 µl) adjusted to the desired pH-value by addition of 2 M NaOH. Benzylmercaptan (40 µl) was added and the mixture was stirred until the starting material was completely consumed. The corresponding C-terminal peptide (100 µl of 10 mM solution) was added and the ligation was monitored by analytical reversed phase HPLC. Gradient: 0–5 min, 5% B; 5–15 min, 5 → 20% B; 15–20 min, 20 → 50% B; flow 1 ml min−1. The eluted peptides were identified by coupled electrospray mass spectrometry. ESI-MS m/z: calcd. for Leu-Tyr-Lys-Ala-Gly-COSBn (25), C33H48N6O6S [M + H]: 657.3, found: 657.3; calcd. for Cys-Arg-Ala-Glu-Tyr-Ser (27), C29H46N9O11S [M + H]: 728.3, found 728.4; calcd. for Isocys-Arg-Ala-Glu-Tyr-Ser (16), C29H46N9O11S [M + H]: 728.3, found: 728.4; calcd. for Leu-Tyr-Lys-Ala-Gly-Cys-Arg-Ala-Glu-Tyr-Ser (28), C55H86N15O17S [M + H]: 1260.6, found: 1260.5; calcd.; for Leu-Tyr-Lys-Ala-Gly-Isocys-Arg-Ala-Glu-Tyr-Ser (26), C55H86N15O17S [M + H]: 1260.6, found: 1260.4.Determination of the pH–rate profile
Aliquots (50 µl) of the ligation mixture were diluted with 20% TFA (30 µl) and analysed by RP-HPLC. The extinction coefficients at 280 nm of 16, 25 and 27 were equal as judged by the peak areas obtained upon HPLC-analysis of equimolar mixtures. The extinction coefficient of the ligation products 26 and 28 was calculated as the sum of the extinction coefficient of 16 and 25 or 25 and 27, respectively. These estimations were reasonable since a) extinction coefficients at 280 nm are determined by the Tyr-absorbance with one tyrosine in 16, 25, 27 and two tyrosine residues in the ligation products 26 and 28, b) hydrolysis of the benzylmercaptan thioester 25 furnished a product which showed an unchanged peak area, thereby confirming that the benzylmercaptan is a weak chromophore at 280 nm. The peak areas corresponding to 16, 25, 26, 27, and 28 were therefore calibrated by weighing factors of 1, 1, 0.5, 1, 0.5, respectively. The relative content of the ligation products was determined and plotted against the reaction time. The initial rates were calculated by determining the slope of the linear range of the resulting curve. A plot of the initial rates of Cys- and Isocys-mediated ligations against the pH-value furnished the pH–rate profile depicted in Fig. 3.References
- D. M. Coltart, Tetrahedron, 2000, 56, 3449–3491 CrossRef CAS.
- P. E. Dawson and S. B. H. Kent, Annu. Rev. Biochem., 2000, 69, 923–960 CrossRef CAS.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776–779 CAS.
- C. F. Liu and J. P. Tam, J. Am. Chem. Soc., 1994, 116, 4149–4153 CrossRef CAS.
- C. F. Liu, C. Rao and J. P. Tam, J. Am. Chem. Soc., 1996, 118, 307–312 CrossRef CAS.
- J. A. Robl, C. Q. Sun, J. Stevenson, D. E. Ryono, L. M. Simpkins, M. P. Cimarusti, T. Dejneka, W. A. Slusarchyk, S. Chao, L. Stratton, R. N. Misra, M. S. Bednarz, M. M. Asaad, H. S. Cheung, B. E. Abboa-Offei, P. L. Smith, P. D. Mathers, M. Fox, T. R. Schaeffer, A. A. Seymour and N. C. Trippodo, J. Med. Chem., 1997, 40, 1570–1577 CrossRef CAS.
- W. T. Ashton, C. L. Cantone, R. L. Tolman, W. J. Greenlee, R. J. Lynch, T. W. Schorn, J. F. Strouse and P. K. S. Siegel, J. Med. Chem., 1992, 35, 2772–2781 CrossRef CAS.
- N. Fotouhi, A. Lugo, M. Visnick, L. Lusch, R. Walsky, J. W. Coffey and A. C. Hanglow, J. Biol. Chem., 1994, 269, 30227–30231 CAS.
- T. Kimmerlin, D. Seebach and D. Hilvert, Helv. Chim. Acta, 2002, 85, 1812–1826 CrossRef CAS.
- L. M. Gustavson and A. Srinivasan, Synth. Commun., 1991, 21, 221–225.
- L. M. Gustavson, D. S. Jones, J. S. Nelson and A. Srinivasan, Synth. Commun., 1991, 21, 249–263.
- Enantioselective synthesis of D- and L-thiomalic acid: B. C. Chen, M. S. Bednarz, O. R. Kocy and J. E. Sundeen, Tetrahedron: Asymmetry, 1998, 9, 1641–1644 Search PubMed.
- M. Schnölzer and S. B. H. Kent, Science, 1992, 256, 221–225 CAS.
- B. Bader, K. Kuhn, D. J. Owen, H. Waldmann, A. Wittinghofer and J. Kuhlmann, Nature, 2000, 403, 223–226 CrossRef CAS.
- H. F. Gaertner, K. Rose, R. Cotton, D. Timms, R. Camble and R. E. Offord, Bioconjugate Chem., 1992, 3, 262–268 CrossRef CAS.
- B. L. Nilsson, L. L. Kiessling and R. T. Raines, Org. Lett., 2000, 2, 1939–1941 CrossRef CAS.
- E. Saxon, J. I. Armstrong and C. R. Bertozzi, Org. Lett., 2000, 2, 2141–2143 CrossRef CAS.
- G. A. Lemieux and C. R. Bertozzi, Trends Biotechnol., 1998, 16, 506–513 CrossRef CAS.
- G. J. Cotton and T. W. Muir, Chem. Biol., 1999, 6, 247–256 CrossRef CAS.
- T. Wieland, E. Bokelmann, L. Bauer, H. U. Lang and H. Lau, Liebigs Ann. Chem., 1953, 583, 129–149 Search PubMed.
- Isocysteine-peptides were observed as by-products in the reaction of a thiocarboxylic acid with β-bromoalanine, which presumably proceeded through an aziridine that was opened to form intermediate 22. J. P. Tam, Y. A. Lu, C. F. Liu and J. Shao, Proc. Natl. Acad. Sci. USA, 1995, 92, 12485–12489 Search PubMed.
- D. Seebach and J. L. Matthews, Chem. Commun., 1997, 2015–2022 RSC.
- R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219–3232 CrossRef CAS.
- P. E. Dawson, M. J. Churchill, M. R. Ghadiri and S. B. H. Kent, J. Am. Chem. Soc., 1997, 119, 4325–4329 CrossRef CAS.
- L. E. Canne, S. J. Bark and S. B. H. Kent, J. Am. Chem. Soc., 1996, 118, 5891–5896 CrossRef CAS.
- J. Offer and P. E. Dawson, Org. Lett., 2000, 2, 23–26 CrossRef CAS.
- T. Kawakami, K. Akaji and S. Aimoto, Org. Lett., 2001, 3, 1403–1405 CrossRef CAS.
- P. Botti, M. R. Carrasco and S. B. H. Kent, Tetrahedron Lett., 2001, 42, 1831–1833 CrossRef CAS.
- C. Marinzi, S. J. Bark, J. Offer and P. E. Dawson, Bioorg. Med. Chem., 2001, 9, 2323–2328 Search PubMed.
- A. J. Kennan, V. Haridas, K. Severin, D. H. Lee and M. R. Ghadiri, J. Am. Chem. Soc., 2001, 123, 1797–1803 CrossRef CAS.
- S. Yao, I. Ghosh, R. Zutshi and J. Chmielewski, Angew. Chem., Int. Ed., 1998, 37, 478–481 CrossRef CAS.
- A. Mattes and O. Seitz, Angew. Chem., Int. Ed., 2001, 40, 3178–3181 CrossRef CAS.
- A. Mattes and O. Seitz, Chem. Commun., 2001, 2050–2051 RSC.
- R. J. Hondal, B. L. Nilsson and R. T. Raines, J. Am. Chem. Soc., 2001, 123, 5140–5141 CrossRef CAS.
- J. W. Haefele and R. W. Broge, Kosmet. Parfüm Drogen Rundsch., 1961, 8, 1–4 Search PubMed.
- The pKa-values were calculated on http://ibmlc2.chem.uga.edu/sparc/..
| This journal is © The Royal Society of Chemistry 2004 |