Ferrocene derivatives as receptors to explore ammonium cation–π interactions
Jiaxin Hua, Leonard J. Barbourb and George W. Gokel*a
aDepartments of Molecular Biology & Pharmacology and Chemistry, Division of Bioorganic Chemistry, Washington University School of Medicine, Campus Box 8103, 660 S. Euclid Ave., St. Louis, MO 63110, USA. E-mail: ggokel@molecool.wustl.edu; Fax: +1 314/362-9298; Tel: +1 314/362-9297
bDepartment of Chemistry, University of Stellenbosch, 7602, Matieland, South Africa
First published on 16th June 2004
Abstract
The potential importance in biology of ammonium–arene cation–π interactions has fostered the development of a model system that uses ferrocene as a scaffold that can rotate groups with respect to each other while maintaining them at a fixed distance. The preparation of 1-benzyl-1′-N,N-dimethylaminomethylferrocene methiodide is reported along with the solid state structure of it and its precursor. Clear evidence is presented for an intermolecular ammonium–arene interaction. The results are analyzed in the context of existing arene–ammonium ion contacts.
Introduction
Cation–π interactions are of enormous potential importance in biological systems where one in every 11 amino acid sidechains is the π-donor benzene, phenol, or indole. These are the respective aromatic residues of the amino acids phenylalanine (Phe, F), tyrosine (Tyr, Y), and tryptophan (Trp, W). Pioneering work by Stucky and coworkers showed that lithium metal reduction of naphthalene led to arene-bound lithium cations.1 Potassium cation interactions with benzene were demonstrated by Kebarle and coworkers in 1981 by using mass spectrometry.2 More recently, several labs have developed model systems3–5 to study cation–π interactions in solution. The field was reviewed in 1997.6 Important work in this area has continued to appear regularly.4,5,7–18We have long been interested in the problem of defining the cation–π interaction specifically for the alkali metal cations Na+ and K+ because these are the most abundant metal cations in vivo. Solution and solid state work that we conducted in the 1980s failed to provide definitive evidence for interactions of these ions with either Na+ or K+ when the π-donors were benzene, alkene, or alkyne.19,20 The latter studies were conducted with lariat ether receptor molecules. Remarkably, minor alterations to the sidechains of these earlier systems21,22 led to a large number of solid state structures that clearly show cation–π interactions. These involve sodium and potassium cations with alkenes,23 alkynes,24 benzene, phenol,25 and indole.26,27 Aromatic residues such as imidazole (the histidine sidechain arene)28 and pentafluorophenyl,29 which were expected not to form π-complexes, did not show cation–π interactions under similar circumstances. We have recently reviewed these results.30,31
Ammonium ion–π complexes are also of great potential interest in biology. The π-donor sidechains of the amino acids are as above. The potential cation–π interaction would involve the sidechain residues of either lysine (Lys, K) or arginine (Arg, R), i.e. amino or guanidine. Note that lysine and arginine occur to the extent of about 1 in 9 amino acids in all known protein sequences. Both the lysine amino group (pKa 10.5) and arginine's guanidine (pKa 12.8) are fully protonated at physiological pH (∼7.4). Thus, cation–π interactions could occur involving the sidechain residues of Lys or Arg with the π-donors Phe, Tyr, or Trp by either interchain or intrachain proximity. The possibility of such interactions was recognized two decades ago by Burley and Petsko, who searched the Protein Data Bank for indications of proximity between sidechain amino groups and arenes.32 Ammonium ion interactions with arenes were also studied using this technique.33 We report here the development of a chemical model system designed to probe such ammonium ion–arene interactions.
Results and discussion
The lariat ether system that we employed successfully to study alkali metal cation–π interactions could be used to complex ammonium ion, NH4+, but not substituted ammonium cations.34 Further, lariat ethers could not be employed for the study of guanidinium cation unless rather large macrocyclic rings were used.35 The latter was not an appealing prospect as studies with alkali metal cations showed that the sidearm's position within the receptor could be critical to the success of π-donor interactions.27 We therefore turned our consideration to other structural scaffolds. It was essential that the scaffold could enforce proximity between cation and π-donor without restricting the interaction. An attractive possibility was the organometallic sandwich complex ferrocene.36Our experience using ferrocene as a scaffold that can undergo rotation on its “molecular ball bearing”37,38 suggested its use in this application. Ferrocene (1) has several useful and unusual properties in the context of supramolecular chemistry. The two aromatic rings of ferrocene are parallel and separated by 3.25 Å. They rotate with respect to each other on an iron atom—a low energy “ball bearing.” Synthetic access is gained readily by acylation of ferrocene's electron rich arenes. Although its use in the present work was not anticipated, the potential for redox switching37 and metal ion donation39 by ferrocene enhanced our interest in this scaffold. After our own effort in this area began, a report appeared in which ferrocene was used as a β-turn mimetic.40 The latter studies were not directed to cation–π interactions, but the use of ferrocene to enforce sidechain proximity is similar in concept to our own approach. An earlier paper reported that ferrocene, when 1,1′-disubstituted by two valine esters, formed intramolecular hydrogen bonds that stabilized a syn sidearm conformation.41
Corey–Pauling–Koltun (CPK) molecular models suggested that when a π-donor and an ammonium cation were present on the 1- and 1′-rings of ferrocene, they could rotate into proximity if such an interaction was favorable. This is illustrated in Scheme 1 for 1-benzyl-1′-trimethylammoniomethylferrocene, 2. Compound 2 was designed to exploit ferrocene's rotational dynamics in the hope that evidence for an ammonium–π interaction would be obtained. The syn and anti conformations of 2 represent the two structural extremes for this compound. When anti, there should be no intramolecular ammonium–π interaction. In the syn conformation, such an interaction might be detectable either in solution or by X-ray methods.
 | ||
| Scheme 1 | ||
In designing 2, we searched the Cambridge Structural Database (CSD) to guide our choice of molecules. Two solid state structures of interest are 1,1′-dibenzylferrocene (CSD: NUYQAZ)42 and 1,1′-bis(2-pyridinomethylaminocarbonyl)ferrocene (JATPUP)43 (Fig. 1). Dibenzylferrocene (3) is in an extended conformation in which the two cyclopentadiene rings are anti and the sidechains are pointed in opposite directions. The structure of 3 has been rendered using the program X-Seed44–46 such that ferrocene is in the space-filling (CPK) metaphor and the chains are represented as sticks. This was done to show the extent to which the cyclopentadienyl rings envelop Fe(II).
 | ||
| Fig. 1 1,1′-Disubstituted ferrocenes 3 and 4 are in conformations in which the sidearms are, respectively, anti and syn. | ||
The structure of picolinamine derivative 4 has two arms, as does 3, but they are positioned on the same side of ferrocene. The organometallic residue is in the syn arrangement and the sidechains exhibit N–H⋯N hydrogen bonding. The latter example supports the notion that when a weak force interaction occurs in the sidechain, the conformation of ferrocene will be appropriate to sustain it. The structures are shown in Fig. 1.
Synthesis of 2
Our initial strategy was to acylate ferrocene (1) with benzoyl chloride and to use the deactivating effect of the acylated ring to direct a Mannich condensation to the (opposite) 1′-ring. The well-known benzoylation proceeded uneventfully but the Mannich condensation on C5H5FeC5H4CO–C6H5 failed to afford the aminomethylated product. An alternate attempt involved benzoylation, as above, followed by treatment of benzoylferrocene with N,N-dimethylcarbamoyl chloride and aluminium chloride. Only starting materials were recovered in this reaction.The successful approach to 2 is shown in Scheme 2. Ferrocene was treated with N,N-dimethylcarbamoyl chloride [(CH3)2NCOCl] and AlCl3 at ambient temperature for 20 h. N,N-Dimethylferrocenecarboxamide 5
(obtained as yellow needles, 42%) was acylated with PhCOCl (2 equiv) and AlCl3
(3 equiv). 1-Benzoyl-1′-N,N-dimethylferrocenecarboxamide (6, C6H5–CO–C5H4FeC5H4–CONMe2) was obtained in 62% yield as a red oil. The latter proved to be unstable and partially decomposed during flash column chromatography. In subsequent preparations, it was reduced immediately with LiAlH4–AlCl3 to afford 7, 1-benzyl-1′-N,N-dimethylaminomethylferrocene (88%), as a red oil that solidified after lengthy drying under high vacuum. Evaporation of a hexane solution containing 7 at 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C for 3 weeks afforded yellow parallelepipeds, mp 48–49°C. These crystals proved to be useful for X-ray analysis. Finally, compound 7 was treated with excess CH3I overnight to afford the ammonium salt 2 as a yellow solid in 90% yield. Crystallization from acetone gave 2 as light yellow rhombohedroids, mp=172–174°C.
°C for 3 weeks afforded yellow parallelepipeds, mp 48–49°C. These crystals proved to be useful for X-ray analysis. Finally, compound 7 was treated with excess CH3I overnight to afford the ammonium salt 2 as a yellow solid in 90% yield. Crystallization from acetone gave 2 as light yellow rhombohedroids, mp=172–174°C.
 | ||
| Scheme 2 | ||
Solid state structure of 7
Single crystal X-ray structures of compounds 2 and 7 have been obtained by direct methods. The structure of dimethylaminoferrocene derivative 7 is shown in Fig. 2. Ferrocene's cyclopentadienide rings and their attached sidearms are in the anti arrangement. It is clear that there is no intramolecular interaction and none was expected. There is, however, a molecule of water that H-bonds between amino nitrogens in adjacent molecules. The bridging water molecule forms an O–H⋯N bond that is (dO–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ) 2.85 Å in length and the O–H⋯N bond angle is nearly linear, i.e. 170°. The distance between nitrogens in adjacent molecules is 5.9 Å and the centroid-to-centroid distance between adjacent benzene rings is essentially the same. The interplane distance of the cyclopentadienes in ferrocene is 3.33 Å and the Cp rings are parallel. No interaction between the sidearms is apparent.
) 2.85 Å in length and the O–H⋯N bond angle is nearly linear, i.e. 170°. The distance between nitrogens in adjacent molecules is 5.9 Å and the centroid-to-centroid distance between adjacent benzene rings is essentially the same. The interplane distance of the cyclopentadienes in ferrocene is 3.33 Å and the Cp rings are parallel. No interaction between the sidearms is apparent. | ||
| Fig. 2 Solid state structure showing two molecules of 7 linked by a water molecule. | ||
Although the structure of precursor 7 exhibits some interesting features, the goal was to detect ammonium ion cation–π interactions in 2. We surmised that the most favorable cation–π interaction would be intramolecular. The structure of compound 2 is shown in Fig. 3. The asymmetric unit consists of two separate molecules in different conformations. In one molecule, the benzene ring turns upward from the ferrocene ring plane at an angle of 107°, as in the structure of 7. The other monomer is disordered. The benzyl sidearm in the latter has two possible directions while the lower part of the ferrocene is fixed. The two arrangements are shown in the left hand structure of Fig. 3. In neither position is there proximity to the benzyl group, and the intramolecular trimethylammonium cation is essentially vertical to the ferrocene unit. The trimethylammonium sidearms adopt similar positions in the two molecules—they turn downward from the ferrocene ring plane at an angle of 111°. The iodide anions are 4.47 Å and 4.66 Å distant from the nearer nitrogen. The sidearms in neither structure are in the anti arrangement. Instead, the sidearms are roughly on the same side of the ferrocene in each case but the dihedral angles are 105°, 69°, and −63°. There does not appear to be any intramolecular cation–π interaction.
 | ||
| Fig. 3 Structure of 1′-benzylferrocenylmethyltrimethylammonium iodide, 2, showing two molecules present in the unit cell. The left hand structure is disordered. | ||
Fig. 4 shows the intermolecular cation–π interaction of three molecules of 2—contributed by two unit cells. The dotted lines indicate contacts between benzene and ammonium ions. The trimethylammonium cation of the central ferrocene is almost vertical at the top of the nearby benzene ring. The nearest methyl carbon is 4 Å away from the benzene centroid. The ammonium cation (N+) is 4.66 Å from the centroid. A second, similar contact occurs in an adjacent pair. A line dropped from nitrogen to the arene's centroid makes an angle of about 12° from vertical. The distance between the cationic nitrogen atom and the arene's centroid is 4.7 Å.
 | ||
| Fig. 4 Three molecules of 2 showing ammonium ion to arene contact. Note the disorder in the right and left monomers. | ||
Compounds exhibiting close ammonium-arene contacts
A search of the CSD revealed five compounds that have close (≤4.7 Å) contacts between a benzene centroid and a quaternary nitrogen atom. Structural drawings of four of these are shown in Fig. 5. Each structure is identified by its CSD descriptor, namely GOFGOX,47 GOVSOZ,48 HIBMEK,49 and WOLWAV.50 Three of the four are organometallic complexes and another three of four involve interactions between different molecules, one of which contains the arene and one that incorporates the ammonium ion. The only intramolecular example is the platinum complex WOLWAV. The distance between quaternary nitrogen and the proximate arene is ∼4.4 Å but a line from nitrogen to the centroid is 23° from perpendicular. Even so, the contact is short and the interaction is therefore significant. | ||
| Fig. 5 Systems having close quaternary ammonium to arene contacts, labeled by their CSD descriptors: GOFGOX, GOVSOZ, HIBMEK, and WOLWAV. | ||
The intermolecular contacts in GOFGOX, GOVSOZ, and HIBMEK are likewise short, all being approximately 4.4 Å. In GOVSOZ, however, the closest benzene ring is angled significantly and the closest carbon is ∼4.23 Å from the nitrogen atom, but the centroid is ∼4.8 Å away. The nitrogen atom of tetra-n-butylammonium ion in HIBMEK is ∼4.2 Å from the nearest phenoxy ring in the bismuth complex shown. The interaction is along a line only 17° from perpendicular. A special situation arises for GOFGOX, in which benzene rings are both inter- and intramolecular with respect to the ammonium nitrogen atom. The ammonium salt's arene is folded back so that there is contact between it and its intramolecular ammonium ion. In addition, the ammonium nitrogen is proximate to the benzene rings present in the resorcinarene. All distances are in the range 4.5–4.7 Å.
An ammonium–π interaction in a substituted ferrocene
The most closely related example that we can find in the literature is a compound (8, Fig. 6) reported as part of an effort to develop compounds that mimic hexesterol, hydrogenated diethylstilbesterol.51 Indeed, this is the only example of an intramolecular contact in a ferrocene that we have found. In this study, one of the benzene rings was replaced by ferrocene. The structure of this compound is reported in the aforementioned paper and appears in the Cambridge Structural Database as LIFZAX. Our own measurement of its nitrogen to centroid distance is 4.70 Å. Note that the arene and ammonium ion interaction is intramolecular rather than intermolecular and the interacting residues are on the same cyclopentadienyl ring, unlike the situation for 2. | ||
| Fig. 6 Solid state structure of 8 as reported in the literature.51 | ||
The contact between an arene and trimethylammonium is necessarily limited in distance by the methyl group hydrogens. This is shown in Fig. 7. Tetramethylammonium cation is shown in panels A and B in the ball and stick and space filling metaphors, respectively. The solvent accessible surface, which reflects the closest approach of a π-donor, is shown in panel C. In principle, the best contact is the one that has both the shortest distance and the most favorable orientation. The ideal interaction between benzene and a point charge would be along a line perpendicular to the aromatic ring. In their study of protein structures, Burley and Petsko found that “[t]he normalized frequency distribution of 1556 amino–aromatic contact distances… reaches a maximum at about 4.75 Å.”32
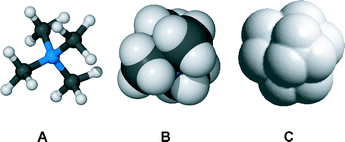 | ||
| Fig. 7 Molecular models of tetra-n-butylammonium cation in (A) the ball and stick and (B) space-filling metaphors. Model C shows the calculated solvent exposed surface. | ||
The findings of Burley and Petsko with respect to bond angle were less clear. The intermolecular interaction between two molecules of 2 can be determined from the crystal structure data. A line drawn from the centroid of one molecule of 2 to the nitrogen of a second molecule is 4.70 Å. The line diverges from perpendicular by ∼19°. It is currently unclear how packing forces influence the angle but the intermolecular π–N+ distance is clearly appropriate for a cation–π contact.
Conclusion
The search for cation–π interactions is both of great current interest and of the profoundest potential importance in biology. The receptor system that is presented here was designed to probe cation–π interactions and does so. In principle, the cation–π interaction observed with 2 could either be intra- or intermolecular. For compound 2, the significant interaction that is observed is between the benzene ring of one molecule and the trimethylammonium ion of a second. The observed bond distance and angle comport with literature values. In principle, this system may be used for more subtle analyses of these interactions and such studies are under way.Experimental
1H-NMR spectra were recorded at 300 MHz in CDCl3 unless otherwise specified. Chemical shifts are reported in ppm. Infrared spectra were recorded on a Perkin-Elmer 1310 infrared spectrophotometer and were calibrated against the 1601 cm−1 band of polystyrene. Melting points were determined on a Thomas-Hoover apparatus in open capillaries and are uncorrected. Thin layer chromatographic (TLC) analyses were performed on silica gel HLO F-254 (0.25 mm thickness), Scientific Adsorbents, Inc. Combustion analyses were performed by Atlantic Microlab, Inc., Atlanta, GA, and are reported as percents. Commercially available solvents and salts were used without further purification.Ferrocene, 1, was obtained commercially and crystallized from heptane prior to use.
1-Benzyl-1′-N,N-dimethylaminomethylferrocene methiodide, 2
Compound 7 (see below, 0.20 g, 0.6 mmol) and CH3I (1 mL) were mixed in 10 mL CH2CI2 and stirred at room temperature overnight. The reaction mixture was concentrated, washed with ether, and maintained under high vacuum for 16 h, during which time the residue solidified. Crystallization from acetone afforded 2 (0.26 g, 90%) as an orange solid, mp 172–174°C. 1H-NMR (CDCI3): 3.21 (s, 9H, –CH3×3), 3.72 (s, 2H, –CH2Ph), 4.21–4.28 (m, 6H, ferrocene), 4.48 (m, 2H, ferrocene), 4.64 (s, 2H, –CH2NME3), 7.18–7.28 (m, 5H, phenyl). 13C-NMR: 35.40, 52.47, 66.93, 69.14, 69.94, 71.29, 72.08, 72.68, 89.67, 126.04, 128.33, 128.69, 141.29. Anal. Calcd for C21H26NIFe: C, 53.10; H, 5.47; N, 2.95%. Found: C, 53.39; H, 5.49; N, 3.05%.Crystal data for 2
Molecular formula: C21H26NIFe; M=475.18, 0.35×0.30×0.20 mm3, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (No. 2), a=9.7909(15), b=13.640(2), c=15.018(2)
Å, α=94.261(3), β=92.364(3), γ=97.445(3)°, V=1980.5(5)
Å3, Z=4, Dc=1.594 g cm−3, F000=952, MoKα radiation, λ=0.71073 Å, T=173(2)K, 2θmax=54.2°, 12560 reflections collected, 8549 unique (Rint=0.0401). Final GooF=1.279, R1=0.1182, wR2=0.2179, R indices based on 5885 reflections with I>2σ(I)
(refinement on F2), 370 parameters, 243 restraints. Lp and absorption corrections applied, μ=2.322 mm−1.
(No. 2), a=9.7909(15), b=13.640(2), c=15.018(2)
Å, α=94.261(3), β=92.364(3), γ=97.445(3)°, V=1980.5(5)
Å3, Z=4, Dc=1.594 g cm−3, F000=952, MoKα radiation, λ=0.71073 Å, T=173(2)K, 2θmax=54.2°, 12560 reflections collected, 8549 unique (Rint=0.0401). Final GooF=1.279, R1=0.1182, wR2=0.2179, R indices based on 5885 reflections with I>2σ(I)
(refinement on F2), 370 parameters, 243 restraints. Lp and absorption corrections applied, μ=2.322 mm−1.N,N-Dimethylferrocenecarboxamide, 5
N,N-Dimethylcarbamoyl chloride (4.30 g, 40 mmol) in CH2Cl2 (10 mL) was added dropwise to a suspension of AlCl3 (5.40 g, 40 mmol) in CH2Cl2 (10 mL) while cooling and the mixture stirred for 10 min (ice bath). The clear solution thus obtained was added dropwise to a CH2Cl2 (15 mL) solution of ferrocene (3.72 g, 20 mmol) at 0°C. The reaction mixture was stirred at room temperature for 20 h, poured into ice–water, and extracted with CH2Cl2
(3×20 mL). The organic layers were combined, washed with saturated Na2CO3
(30 mL) and water (30 mL) and then dried over MgSO4. Flash column chromatography (silica gel, eluted with 1 ∶ 4 Me2CO–hexanes) afforded 5 as yellow needles, (2.15 g, 42%) which was recrystallized from hexanes, mp=114–115°C (lit.52 mp=119–121°C).1-Benzoyl-1′-N,N-dimethylferrocenecarboxamide, 6
Benzoyl chloride (0.77 g, 5.5 mmol) in 30 mL CH2Cl2 was added dropwise to a suspension of AlCl3 (1.10 g, 8.1 mmol) in 20 mL CH2Cl2 while cooling to −30°C. N,N-dimethylferrocenecarboxamide (0.70 g, 2.7 mmol) in 20 mL CH2Cl2 was then added dropwise during 10 min. The reaction mixture was stirred at −30°C for 1 h, and at 0°C for 5 h, poured into ice–water, extracted with CH2Cl2
(3×20 mL), the organic layers were combined and washed with saturated Na2CO3
(30 mL), and water (30 mL). After drying (MgSO4), flash column chromatography (silica gel, eluted with 1 ∶ 4 Me2CO–hexanes) afforded 3 as a dark red oil (0.6 g, 62%). IR: 3088, 2927, 1636, 1619, 1499, 1449, 1395, 1376, 1286 cm−1. 1H-NMR (CDCl3): 3.01 (s, 6H, –CH3), 4.33–5.30 (m, 8H, ferrocene), 7.48–7.89 (m, 5H, PhH). 13C-NMR: 36.20, 38.84, 71.42, 72.23, 72.96, 74.74, 78.85, 80.42, 128.06, 128.18, 131.65, 139.33, 169.11, 198.50.1-Benzyl-1′-N,N-dimethylaminomethylferrocene 7
AlCl3 (0.44 g, 3.3 mmol) was dissolved in 20 mL ether and was added dropwise to a suspension of LiAlH4 (0.13 g, 3.3 mmol) in 20 mL ether. Compound 6 (0.60 g, 1.7 mmol) in 10 mL ether was added dropwise to the reductive mixture. After addition, it was refluxed for 2 h. H2O was added to destroy the reductive agents. 30 mL 2 M NaOH solution was added, and the water layer was extracted with ether. The organic layer was dried with MgSO4, concentrated, and flash column chromatography (silica gel, eluted with 1 ∶ 2 Me2CO–hexanes, with 1% Et3N) afforded 7 as a red oil (0.5 g, 88%), which solidified under high vacuum. Crystallization from hexanes afforded yellow crystals, mp=48–49°C. IR: 3084, 3028, 2936, 2812, 2765, 1603, 1495, 1454, 1039, 1021 cm−1. 1H-NMR (CDCl3): 2.15 (s, 6H, –CH3), 3.22 (s, 2H, –CH2NMe2), 3.66 (s, 2H, –CH2Ph), 4.02–4.10 (m, 8H, ferrocene), 7.15–7.25 (m, 5H, phenyl). 13C-NMR: 35.75, 44.67, 58.91, 68.05, 68.70, 69.06, 70.67, 83.25, 87.91, 125.83, 128.15, 128.25, 141.43. Anal. Calcd for C20H23NFe: C, 72.12; H, 6.91; N, 4.21%. Found: C, 72.24; H, 6.82; N, 4.13%.Crystal data for 7
Molecular formula: C40H48Fe2N2O; M=684.50, 0.25×0.25×0.10 mm3, monoclinic, space group C2/c
(No. 15), a=31.690(5), b=5.9141(9), c=19.117(3)
Å, β=110.715(3)°, V=3351.2(9)
Å3, Z=4, Dc=1.357 g cm−3, F000=1448, MoKα radiation, λ=0.71073 Å, T=173(2)K, 2θmax=54.2°, 9145 reflections collected, 3688 unique (Rint=0.0488). Final GooF=0.997, R1=0.0433, wR2=0.0861, R indices based on 2640 reflections with I>2σ(I)
(refinement on F2), 208 parameters, 0 restraints. Lp and absorption corrections applied, μ=0.900 mm−1. CCDC reference numbers 222951–222952. See http://www.rsc.org/suppdata/nj/b3/b313422a/ for crystallographic data in .cif or other electronic format.Acknowledgements
We thank the Petroleum Research Fund, administered by the American Chemistry Society, for a grant (PRF 37197-AC4) that supported this work.References
- J. J. Brooks, W. Rhine and G. D. Stucky, J. Am. Chem. Soc., 1972, 94, 7346–7351 CrossRef CAS.
- J. Sunner, K. Nishizawa and P. Kebarle, J. Phys. Chem., 1981, 85, 1814–1820 CrossRef CAS.
- P. C. Kearney, L. S. Mizoue, R. A. Kumpf, J. E. Forman, A. McKurdy and D. A. Dougherty, J. Am. Chem. Soc., 1993, 115, 9907–9919 CrossRef CAS.
- K. Aoki, K. Maruyama and H. Nishiyama, J. Chem. Soc., Chem. Commun., 1995, 2221–2222 RSC.
- F. Inokuchi, Y. Miyahara, T. Inazu and S. Shinkai, Angew. Chem., Int. Ed. Engl., 1995, 34, 1364–1366 CrossRef CAS.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS.
- O. M. Cabarcos, C. J. Weinheimer and J. M. Lisy, J. Chem. Phys., 1998, 108, 5151–5154 CrossRef CAS.
- K. Murayama and K. Aoki, Inorg. Chim. Acta, 1998, 36–42 CrossRef CAS.
- F. Nachon, L. Ehret-Sabatier, D. Loew, C. Colas, A. van Dorsselaer and M. Goeldner, Biochemistry, 1998, 37, 10507–13 CrossRef CAS.
- S. M. Ngola and D. A. Dougherty, J. Org. Chem., 1998, 63, 4566–4567 CrossRef CAS.
- A. Y. Ting, I. Shin, C. Lucero and P. G. Schultz, J. Am. Chem. Soc., 1998, 120, 7135–7136 CrossRef CAS.
- J. Wouters, Protein Sci., 1998, 7, 2472–2475 CAS.
- W. Zhonge, J. P. Gallivan, Y. Zhang, L. Li, H. A. Lester and D. A. Dougherty, Proc. Natl. Acad. Sci. USA, 1998, 95, 12088–12093 CrossRef CAS.
- O. M. Cabarcos, C. J. Weinheimer and J. M. Lisy, J. Chem. Phys., 1999, 110, 8429–8435 CrossRef CAS.
- B. T. King, B. C. Noll and J. Michl, Collect. Czech. Chem. Commun., 1999, 64, 1001–1012 CrossRef CAS.
- J. B. Nicholas, B. P. Hay and D. A. Dixon, J. Phys. Chem. A, 1999, 103, 1394–1400 CrossRef CAS.
- R. C. Dunbar, J. Phys. Chem. A, 2000, 104, 8067–74 CrossRef CAS.
- D. Feller, Chem. Phys. Lett., 2000, 322, 543–548 CrossRef CAS.
- R. D. Gandour, F. R. Fronczek, V. J. Gatto, C. Minganti, R. A. Schultz, B. D. White, K. A. Arnold, D. Mazzocchi, S. R. Miller and G. W. Gokel, J. Am. Chem. Soc., 1986, 108, 4078–4088 CrossRef CAS.
- K. A. Arnold, A. M. Viscariello, M. Kim, R. D. Gandour, F. R. Fronczek and G. W. Gokel, Tetrahedron Lett., 1988, 3025–3028 CrossRef CAS.
- E. S. Meadows, S. L. De Wall, L. J. Barbour and G. W. Gokel, J. Am. Chem. Soc., 2001, 123, 3092–3107 CrossRef CAS.
- J. Hu, L. J. Barbour and G. W. Gokel, Proc. Natl. Acad. Sci. USA, 2002, 99, 5121–5126 CrossRef.
- J. Hu, L. J. Barbour and G. W. Gokel, Chem. Commun., 2001, 1858–1859 RSC.
- J. Hu, L. J. Barbour and G. W. Gokel, J. Am. Chem. Soc., 2001, 123, 9486–9487 CrossRef CAS.
- S. L. De Wall, L. J. Barbour and G. W. Gokel, J. Am. Chem. Soc., 1999, 8405–8406 CrossRef.
- S. L. De Wall, E. S. Meadows, L. J. Barbour and G. W. Gokel, J. Am. Chem. Soc., 1999, 121, 5613–5614 CrossRef.
- J. Hu, L. J. Barbour and G. W. Gokel, J. Am. Chem. Soc., 2002, 124, 10940–10941 CrossRef CAS.
- J. Hu, L. J. Barbour, R. Ferdani and G. W. Gokel, Chem. Commun., 2002, 1810–1811 RSC.
- S. L. De Wall, E. S. Meadows, L. J. Barbour and G. W. Gokel, Proc. Natl. Acad. Sci. USA, 2000, 97, 6271–6276 CrossRef CAS.
- G. W. Gokel, S. L. D. Wall and E. S. Meadows, Eur. J. Org. Chem., 2000, 2967–2978 CrossRef CAS.
- G. W. Gokel, L. J. Barbour, R. Ferdani and J. Hu, J. Acc. Chem. Res., 2002, 35, 878–86 Search PubMed.
- S. K. Burley and G. A. Petsko, FEBS Lett., 1986, 203, 139–143 CrossRef CAS.
- M. Meot-Ner and C. A. Deakyne, J. Am. Chem. Soc., 1985, 107, 474–479 CrossRef CAS.
- R. A. Schultz, E. Schlegel, D. M. Dishong and G. W. Gokel, J. Chem. Soc., Chem. Commun., 1982, 242–243 RSC.
- K. Madan and D. J. Cram, J. Chem. Soc., Chem. Commun., 1975, 427–8 RSC.
- M. Rosenblum, Chemistry of the Iron Group Metallocenes: Ferrocene, Ruthenocene, Osmocene, Interscience Publishers, New York, 1965 Search PubMed.
- J. C. Medina, I. Gay, Z. Chen, L. Echegoyen and G. W. Gokel, J. Am. Chem. Soc., 1991, 113, 365–366 CrossRef CAS.
- J. C. Medina, C. Li, S. G. Bott, J. L. Atwood and G. W. Gokel, J. Am. Chem. Soc., 1991, 113, 366–367 CrossRef CAS.
- J. C. Medina, T. T. Goodnow, S. Bott, J. L. Atwood, A. E. Kaifer and G. W. Gokel, J. Chem. Soc., Chem. Commun., 1991, 290–292 RSC.
- D. R. van Staveren, T. Weyhermueller and N. Metzler-Nolte, Dalton Trans,, 2003, 210–220 RSC.
- R. S. Herrick, R. M. Jarret, T. P. Curran, D. R. Dragoli, M. B. Flaherty, S. E. Lindyberg, R. A. Slate and L. C. Thornton, Tetrahedron Lett., 1996, 37, 5289–5292 CrossRef CAS.
- J. J. Byrne, A. Rebello, P. Y. Chavant, M. T. Averbuch-Pouchot and Y. Vallee, Z. Kristallogr. – New Cryst. Struct., 1998, 213, 180 Search PubMed.
- J. D. Carr, S. J. Coles, W. W. Hassan, M. B. Hursthouse, K. M. A. Malik and J. H. R. Tucker, J. Chem. Soc., Dalton. Trans., 1999, 57–62 RSC.
- L. J. Barbour, http://www.x-seed.net/, 2003.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189–191 Search PubMed.
- J. L. Atwood and L. J. Barbour, Cryst. Growth Des., 2003, 3, 3–8 CrossRef CAS.
- K. Murayama and K. Aoki, Chem. Lett., 1998, 301–302 CrossRef CAS.
- T. B. Wen, B. S. Kang, C. Y. Su, D. X. Wu, L. G. Wang, S. Liao and H. Q. Liu, Bull. Soc. Jpn., 1998, 71, 2339–2343 Search PubMed.
- P. Hodge, S. C. James, N. C. Norman and A. G. Orpen, J. Chem. Soc., Dalton Trans., 1998, 4049–4054 RSC.
- G. Ferguson, Y. Li, A. J. McAlees, R. McCrindle and K. Xiang, Organometallics, 1999, 18, 2428–2439 CrossRef CAS.
- B. Malezieux, M. Gruselle, L. L. Troitskaya, V. I. Sokolov and J. Vaissermann, Organometallics, 1994, 13, 2979–2986 CrossRef CAS.
- T. H. Galow, J. Rodrigo, K. Cleary, G. Cooke and V. M. Rotello, J. Org. Chem., 1999, 64, 3745–3746 CrossRef.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2004 |