The crystal structures of three primary products from the selective reduction of 2,4,6-trinitrotoluene
Duncan
Graham
a,
Alan R.
Kennedy
*a,
Callum J.
McHugh
a,
W. Ewen
Smith
a,
William I. F.
David
b,
Kenneth
Shankland
b and
Norman
Shankland
cd
aDepartment of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow G1 1XL, Scotland
bISIS Facility, CLRC Rutherford Appleton Laboratory, Chilton, Oxfordshire, OX11 0QX, England
cDepartment of Pharmaceutical Sciences, University of Strathclyde, 27 Taylor Street, Glasgow G4 0NR, Scotland
dCrystallografX Limited, 38 Queen Street, Glasgow G1 3DX, Scotland. E-mail: a.r.kennedy@strath.ac.uk; Fax: 0141 552 0876; Tel: 0141 548 2016
First published on 22nd October 2003
Abstract
The crystal structures of three primary products from the selective reduction of 2,4,6-trinitrotoluene (TNT) have been determined by synchrotron X-ray powder diffraction (2-amino-4,6-dinitrotoluene) and single crystal X-ray diffraction (4-amino-2,6-dinitrotoluene and 2-hydroxyamino-4,6-dinitrotoluene). The molecular structure of 2-amino-4,6-dinitrotoluene, including rotational disorder of the 6-nitro group, was subsequently detailed to a higher resolution by a single-crystal analysis. In contrast to the known structures of TNT, the crystal structures of these amino species are dominated by hydrogen-bonded sheets connected via ring stacking, whilst that of 2-hydroxyamino-4,6-dinitrotoluene is dominated by the dual hydrogen-bonding acceptor/donator role of the hydroxyamine group.
Introduction
The reduction chemistry of high explosives such as 2,4,6,-trinitrotoluene (TNT) and 1,3,5-trinitro-1,3,5-triazacyclohexane (RDX) is an important area of current research, in particular amongst groups investigating the treatment and clean-up of land contaminated with explosive residues.1 Our own interest in this chemistry is in the detection of trace levels of an explosive’s vapour by Surface Enhanced Resonance Raman Scattering (SERRS). Whilst a lack of sensitivity limits the use of normal Raman scattering in the detection of explosive vapours or trace amounts, SERRS should enable detection of TNT at the levels required. In order to exploit the SERRS effect, the analyte must be adsorbed onto the SERRS metal substrate and should contain a chromophore that absorbs visible wavelengths of light. As neither TNT nor RDX possesses either of these attributes, chemical modification of the explosive is required to obtain a coloured derivative that possesses the functionality needed to enable strong interaction with a SERRS metal substrate. We have recently described the development of synthetic routes to such highly coloured TNT2 and RDX3 derivatives suitable for SERRS analysis and reported on the optimisation of their SERRS response.Herein, we report on the crystal structures of the main amino and hydroxyamino products formed upon initial selective reduction of TNT, 2-amino-4,6-dinitrotoluene [1], 4-amino-2,6-dinitrotoluene [2] and 2-hydroxyamino-4,6-dinitrotoluene [3]. In TNT contaminated environments these reduction products are formed by the action of bacterial nitroreductase and, although they contribute significantly to the toxicity of contaminated soil, they are necessary intermediates en route to the non-toxic and fully reduced 2,4,6-triaminotoluene.1 The molecular recognition properties of these species are thus of interest with regard to their binding to nitroreductase enzymes as well as to soils and metal surfaces.
Experimental
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) <1.5 ppm]. Anal. Calcd for C7H7N3O4:C, 42.64; H, 3.55; N, 21.32. Found C, 42.32; H, 3.21; N, 21.34%.
<0.2 ppm]. Anal. Calcd for C7H7N3O4: C, 42.64; H, 3.55; N, 21.32. Found C, 42.01; H, 3.39; N, 20.78.
×10 mL). The organic layer was dried over sodium sulfate and purified by column chromatography, eluting with dichloromethane to afford 3 as a yellow/orange solid (0.5 g, 38%). Crystallisation from chloroform gave 3 as fine orange needles (0.22 g). Rf
(B) 0.18. 1H NMR (acetone-d6)
δ 2.29 (3H, s, CH3), 8.04 (1H, s, ArH), 8.19 (1H, s, ArH), 8.39 (1H, s, OH), 8.42 (1H, s, NH). m/z
(EI) 213.03817 [C7H7N3O5
(M)+<1.9 ppm]. Anal. Calcd for C7H7N3O5: C, 39.43; H, 3.28; N, 19.72. Found C, 38.02; H, 2.07; N, 18.84.
<1.5 ppm]. Anal. Calcd for C7H7N3O4:C, 42.64; H, 3.55; N, 21.32. Found C, 42.32; H, 3.21; N, 21.34%.
<0.2 ppm]. Anal. Calcd for C7H7N3O4: C, 42.64; H, 3.55; N, 21.32. Found C, 42.01; H, 3.39; N, 20.78.
×10 mL). The organic layer was dried over sodium sulfate and purified by column chromatography, eluting with dichloromethane to afford 3 as a yellow/orange solid (0.5 g, 38%). Crystallisation from chloroform gave 3 as fine orange needles (0.22 g). Rf
(B) 0.18. 1H NMR (acetone-d6)
δ 2.29 (3H, s, CH3), 8.04 (1H, s, ArH), 8.19 (1H, s, ArH), 8.39 (1H, s, OH), 8.42 (1H, s, NH). m/z
(EI) 213.03817 [C7H7N3O5
(M)+<1.9 ppm]. Anal. Calcd for C7H7N3O5: C, 39.43; H, 3.28; N, 19.72. Found C, 38.02; H, 2.07; N, 18.84.
Crystallography
Single crystal measurements on 1, 2 and 3 were carried out on a Nonius Kappa CCD diffractometer using MoKα radiation, λ=0.71073 Å. All non-disordered, non-H-atoms for 2 and 3 were refined anisotropically and the H-atoms bonded to N or O were refined isotropically. All other H-atoms were placed in calculated positions and in riding modes. The structures were refined against F2 to convergence using the SHELXL-97 program.4 Specific crystallographic data and refinement parameters are given in Table 1.†
| 1 | 2 | 3 | |
|---|---|---|---|
| Formula | C7H7N3O4 | C7H7N3O4 | C7H7N3O5 |
| Formula weight | 197.16 | 197.16 | 213.16 |
| Crystal system | Monoclinic | Triclinic | Monoclinic |
| Space group | P21/a |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
| a/Å | 6.7072(5) | 8.1036(3) | 15.0980(4) |
| b/Å | 16.0387(11) | 8.0502(3) | 3.9219(1) |
| c/Å | 8.1083(5) | 14.2752(5) | 16.4446(5) |
| α/° | 75.097(2) | ||
| β/° | 91.160(4) | 74.059(2) | 116.586(2) |
| γ/° | 78.453(5) | ||
| V/Å3 | 872.07(10) | 847.35(5) | 870.77(4) |
| Z | 4 | 4 | 4 |
| T/K | 295 | 150 | 150 |
| 2θ max./° | 50.04 | 54.94 | 57.20 |
| Refl. measured | 7058 | 13402 | 10574 |
| Refl. unique | 1515 | 3812 | 2227 |
| R int | 0.034 | 0.036 | 0.057 |
| R1 | 0.0645 | 0.0611 | 0.0417 |
| wR2 | 0.1835 | 0.1852 | 0.1120 |
| Refl. observed | 1111 | 3221 | 1628 |
| Parameters | 143 | 271 | 145 |
| GoF | 1.100 | 1.122 | 1.042 |
Initial attempts to solve the structure of 1 from single crystal data collected at 123 K failed. The problem was investigated by collecting X-ray powder diffraction (XRPD) data from a polycrystalline sample of 1 contained in a 1 mm capillary at station BM165 of the European Synchrotron Radiation Facility, Grenoble (λ=0.8000 Å). At 105 K the sample was extremely line-broadened, whereas at 293 K the pattern exhibited acceptably sharp diffraction features (see A comparison of single crystal and XRPD analyses of crystal structure 1, below). A full data set was therefore collected at 293 K with a view to solving the crystal structure. An appropriate data collection scheme similar to the one described in ref. 6 was employed in order to increase preferentially the quality of short d-spacing reflections and thus increase the probability of solving the structure. The data were indexed to a monoclinic cell [a=6.703, b=16.031, c=8.106 Å, β=91.034°, V=870.9 Å3] using DICVOL91,7 with promising figures of merit, M(23)=30, F(23)=161. The volume of the unit cell suggested Z=4 and a preliminary examination of the predicted peak positions for this cell against the measured data indicated that the space group was P21/a. Diffraction data in the range 1.5–25.0° 2θ
(equating to ∼1.8 Å resolution) were fitted using the Pawley method in space group P21/a using DASH8 to give a profile χ2 of 7.6, with no significant misfit in the pattern. The crystal structure was then solved using the simulated annealing procedure described previously9 that is now implemented in DASH.
A satisfactory fit to the data could not be obtained with a structural model in which all atoms occupied fully ordered positions. However, the fit to the data improved significantly when the 6-nitro group was treated as being disordered. Each O-atom was allowed to occupy two separate positions with site occupancy factors (SOF’s) constrained to sum to unity. Since the amount of useable diffraction data was insufficient to refine the SOF’s accurately, the structure was solved using different fixed values of the SOF’s and the solution returning the best fit to the data (Table 2, Model 4) was taken to be the most probable solution.
| Model | SOF's | Profile χ2 | Intensities χ2 |
|---|---|---|---|
| 1 | 100∶0 |
41 | 203 |
| 2 | 90∶10 |
36 | 174 |
| 3 | 80∶20 |
33 | 157 |
| 4 | 70∶30 |
32 | 154 |
| 5 | 60∶40 |
33 | 160 |
| 6 | 50∶50 |
34 | 163 |
A single crystal data collection on 1 was subsequently repeated, but this time with the sample temperature held at 293 K. The structure solved and refined satisfactorily to give SOF's for the O-atoms of the 6-nitro group equal to 79.8(8)∶20.2(8), in good agreement with the XRPD data. The solution and refinement details were as for 2 and 3, with the O-atoms of the minor disordered component refined isotropically.
Results and discussion
Molecular structures
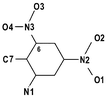
As in the known structures of TNT10 and other nitro-aromatics,11 the major deviations from idealised aromatic geometry in the molecular structures of 1, 2 and 3 include a marked widening of the internal ring angles at the C-atoms bonded to nitro groups, with angles ranging from 123.3(2)° to 125.8(2)°. Of the other internal angles, only the angle at the C-atom bonded to the hydroxyamine group in 3 (=121.28(13)°) is significantly greater than 120°; the remaining angles tend to be equal to or less than 120°, reaching a minimum of 111.6(2)° at C1 in 2
(Table 3).
| 1 | 2 | 3 | ||
|---|---|---|---|---|
|
a SOF(O4)=79.8(8)
|
||||
|

|

|
||
| C2-C1-C6 | 116.5(2) | 111.7(2) | 111.6(2) | 116.1(1) |
| C2-C1-C7 | 118.3(2) | 124.0(2) | 124.3(2) | 119.9(1) |
| C6-C1-C7 | 125.1(2) | 124.1(2) | 123.9(2) | 124.0(1) |
| C1-C2-C3 | 119.4(2) | 125.6(2) | 125.7(2) | 121.3(1) |
| C1-C2-N1 | 120.6(2) | 120.0(2) | 119.8(2) | 116.6(1) |
| C3-C2-N1 | 120.0(2) | 114.4(2) | 114.6(2) | 122.1(1) |
| C2-C3-C4 | 119.9(2) | 119.8(2) | 119.9(2) | 118.3(1) |
| C3-C4-C5 | 123.3(2) | 117.3(2) | 117.2(2) | 123.4(1) |
| C3-C4-N2 | 118.3(2) | 121.3(2) | 120.9(2) | 118.3(1) |
| C5-C4-N2 | 118.4(2) | 121.4(2) | 121.9(2) | 118.2(1) |
| C4-C5-C6 | 115.6(2) | 120.1(2) | 119.8(2) | 115.7(1) |
| C5-C6-C1 | 125.3(2) | 125.6(2) | 125.8(2) | 125.1(1) |
| C5-C6-N3 | 115.2(2) | 114.6(2) | 114.9(2) | 116.2(1) |
| C1-C6-N3 | 119.5(2) | 119.9(2) | 119.2(2) | 118.6(1) |
| C7C1C2N1 | 0.7(4) | 8.5(3) | −9.5(4) | 1.9(2) |
| C7C1C6N3 | −2.6(5) | −8.5(3) | 12.1(4) | 4.0(2) |
| C3C4N2O2 | 178.4(3) | — | — | −2.9(2) |
| C1C2N1O1 | — | 36.3(3) | −34.6(3) | −169.7(1) |
| C1C6N3O4 | −51.2(5)a | −38.2(3) | 38.2(3) | 54.6(2) |
These marked distortions from the ideal sp2 geometry, ranging from 111.6(2)° to 125.8(2)°, are in fact entirely in keeping with expectations based on Domenicano's assessment12 of structural substituent effects in benzene derivatives. Following Domenicano and utilising the angular substituent parameters for –CH3, –NO2and –NH2,12 it can be seen that ten of the twelve independent pairs of predicted and observed internal ring angles in 1 and 2 agree to within three e.s.d's (≈1°). The remaining two observed angles (at C1 and C4 in 2) are both 1.4° narrower than predicted. This discrepancy may be due to a “push–pull” effect with substituents interacting cooperatively through the aromatic ring, such that the overall observed effect is greater than the expected sum of parts.12,13 Similar predictions for angular deviations in 3 are not possible as a search of the Cambridge Structural Database14 found no examples of aromatic hydroxyamines. However, a useful comparison can be made between 1 and 3. Replacing NH2 in 1 with NHOH in 3 causes the internal angle at C2 to widen by 1.9° and the angle at C3 to narrow by 1.6° (the other internal angles agree to within two e.s.d’s). This suggests that the NHOH group has considerably less electron-donating character than NH2,12 as would be expected on simple chemical grounds.
Compound 2 has a non-crystallographic mirror plane running through C7C1C4N2 in Fig. 1, which is rendered to emphasise the non-coplanarity of the nitro groups to the aromatic ring. In 1 and 3 (Fig. 2), the nitro groups ortho to the methyl group are similarly twisted out of the ring plane, while the nitro groups para to methyl are essentially coplanar with the ring (Table 3). This difference is attributable to steric interaction between adjacent methyl and nitro groups. An interesting consequence of the distorted geometry at C1 in 2 is the fact that the methyl C-atom is displaced out of the ring plane by a significant amount in each independent molecule (0.175(3) Å and 0.145(3) Å cf. 0.024(4) Å in 1 and 0.039(2) Å in 3).
 | ||
| Fig. 1 The structure of one of the crystallographically independent molecules of 2, with ellipsoids rendered at 50% probability. | ||
 | ||
| Fig. 2 The structure of 3, with ellipsoids rendered at 50% probability. | ||
Intermolecular interactions
The crystal packing of TNT is of considerable significance as it is perceived to govern the subtle and complex relationships between TNT polymorphs that impact upon the explosive's stability and use in the solid-state.10 In 1, 2 and 3 we have replaced one of TNT's weakly hydrogen-bond accepting nitro groups with a strong hydrogen-bond donating group. The consequence of this is not only to introduce a stronger hydrogen-bonding network, but also that all three new structures display face-to-face ring stacking not seen in TNT. Both amino compounds 1 and 2 form layer structures with extensive hydrogen-bonding (both N–H⋯O and (ar)C–H⋯O interactions) that occurs exclusively within the layers (Fig. 3). Compound 1 shows a simple sheet structure with all layers mutually parallel and perpendicular to the a axis. However 2, which has two molecular conformations A and B per asymmetric unit, has a more complicated AABB layering with each layer approximately perpendicular to the c axis. The molecules of the neighbouring A and B sheets (Fig. 3) are roughly parallel in contrast to the anti-parallel arrangement found across the double layers AA and BB. | ||
| Fig. 3 A section of the hydrogen-bonded B layer of 2. | ||
In both 1 and 2 these internally hydrogen-bonded layers are bound to the next layer by offset face-to-face π-stacking. 3 is of a markedly different structural type (Fig. 4). A simple translation along b gives stacks of aromatic rings with a large face-to- face spacing of 3.922 Å (cf. the much shorter face-to-face distances (3.3–3.5 Å) in 1 and 2). Adjacent stacks along the c direction are tilted with respect to each other and are bound into pairs of stacks by the strong O–H⋯O hydrogen-bonds that zig-zag between neighbouring hydroxy groups. The amine H-atom forms a similarly patterned, but longer range, connection to the 4-nitro group of a second neighbouring stack.
 | ||
| Fig. 4 Crystal packing in 3 viewed along the b axis. | ||
Given the polynitro nature of TNT and its derivatives reported herein, we expected to find evidence of nitro–nitro O⋯N contacts as described by Wozniak et al.15 However, upon examination of the twenty independent nitro groups of TNT and its derivatives, only those in 2 had O⋯N contacts shorter than (the formally repulsive) O⋯O or O⋯C contacts. The closest such contact occurs in 3
(O5⋯O5*=2.806 Å
cf. sum of van der Waals radii=3.04 Å16 to 3.12 Å17), but, even taken as a group, the formally repulsive O⋯O contacts are typically shorter than the formally attractive N⋯O contacts (N⋯O range 3.041 Å to 3.102 Å). This counter-intuitive behaviour of the nitro group was recently noted by Szczęsna and Urbańczyk-Lipkowska,18 who suggested that short O⋯O contacts were favoured by intramolecular resonance assisted hydrogen-bonding (RAHB). This does not seem applicable to the structures of TNT and its derivatives, which have no intramolecular RAHB. In these compounds, an anisotropic van der Waals radius19 for nitro-O seems a plausible mechanism by which to account for unexpectedly short intermolecular O⋯O contacts.
A comparison of single crystal and XRPD analyses of crystal structure 1
The XRPD patterns measured for 1 exhibited much broader peak widths at 105 K than at RT, indicating greater mosiacity. Lattice parameters measured by single crystal diffraction between RT [a=6.703, b=16.031, c=8.106 Å, β=91.034°] and 123 K [a=6.534, b=15.939, c=8.061 Å, β=90.195°] tended to suggest a gradual and reversible monoclinic→orthorhombic transition, with a transition temperature <123 K (123 K was the lowest sample temperature to yield useful data). The percentage thermal contraction is greatest in the a direction, corresponding to a decrease in the distance between the layers in 1. As the disordered 6-nitro group projects out of the layer plane (Fig. 5), it is forced closer to the neighbouring molecular layer at lower temperatures and the resulting steric strain would appear to lead to increased mosiaicity. It is also interesting to speculate that the O-atoms of the 6-nitro group become disordered across two sites in an attempt to alleviate this strain.
 | ||
| Fig. 5 Crystal structure 1, showing the disordered 6-nitro group projecting between parallel sheets of molecules. | ||
A comparison of the XRPD and single-crystal structures (Fig. 6) shows excellent agreement with respect to the position and orientation of the molecule in the unit cell, and the orientation of the 6-nitro group at higher occupancy. Unsurprisingly, the orientation of the lower occupancy 6-nitro group is less well determined. Subsequent structure determinations that included a preferred orientation correction improved the overall fit to the data (with a decrease in profile χ2 from ∼32 to ∼26), but did not alter the rank order of the disordered models (as listed in Table 2) or improve on the orientation of the lower occupancy 6-nitro group.
 | ||
| Fig. 6 The crystal structure of 1 determined from XRPD data (red) overlaid upon the corresponding single-crystal solution (green). Each O-atom of the 6-nitro group is disordered between high occupancy (80% single crystal; 70% XRPD) and low occupancy sites and the two structure solutions are in good agreement on the position of the higher occupancy site. Note that the H-atoms of the 1-methyl group in the XRPD solution have been placed in calculated positions. | ||
Other authors have shown that the results obtained from global optimisation can sometimes indicate the presence of conformational disorder.20,21 Huq and Stephens,21 for example, found that two distinct conformations of ranitidine gave equally good fits to XRPD data obtained from a polycrystalline sample of ranitidine hydrochloride and went on to show that these matched the conformations found in the disordered structure determined by single crystal diffraction. Here, we have taken the approach that residual misfit in the diffraction pattern derived from a fully ordered model might well arise from disorder in the actual structure and have used global optimisation to prove this hypothesis by simultaneously optimising models that include contributions from atoms with fractional occupancies.
Conclusions
In determining the disordered structure of 1 from XRPD data, chemical intuition plays an essential role in identifying the disordered atoms, which can then be correctly incorporated into the structure solution model. This approach is distinct from the more general one of using difference Fourier maps to complete a partial structure solution, the latter being problematic when low-resolution, low quality XRPD data makes map interpretation difficult. The combination of chemical intuition and molecular connectivity, on the other hand, can clearly produce excellent results even at ∼1.8 Å resolution. The XRPD solution agrees well with the solution from single crystal data and discrepancies between the two structures are ascribed simply to the relative amounts of diffraction data; 1111 observed reflections to ∼0.8 Å resolution for the single-crystal structure compared with only 146 reflections to ∼1.8 Å resolution in the XRPD data set.Large deformations from ideal sp2 geometry are observed in the aromatic rings of 1, 2 and 3 and this is entirely in line with expectations based on a knowledge of structural substituent effects in benzene derivatives. The introduction of stronger hydrogen-bond donors to TNT results in solid-state structures with layer architectures as opposed to the herring bone motifs seen in TNT itself. 1 and 2 form hydrogen bonded sheets connected via N–H and C–H hydrogen-bond donation to nitro acceptors. These sheets are held to each other by π-stacking and by dipole–dipole interactions between nitro groups. The stronger hydrogen-bonding in 3 (with its donating and accepting NHOH group) gives a different structure dominated by NHOH–NHOH dimers.
Acknowledgements
We gratefully acknowledge the Home Office UK for financial support for CJM, Sally Price of the UCL Centre for Theoretical and Computational Chemistry for helpful input and the ESRF for providing a beamtime award (CH849).References
- A. Esteve-Nunez, A. Caballero and J. L. Ramos, Microbiol. Mol. Biol. Rev., 2001, 65, 335 Search PubMed; J. D. Rodgers and N. J. Bunce, Environ. Sci. Technol., 2001, 35, 406 CrossRef CAS; H. Y. Kim and H. G. Song, J. Microbiol., 2000, 38, 250 Search PubMed; N. Hannink, S. J. Rosser, C. E. French, A. Basran, J. A. H. Murray, S. Nicklin and N. C. Bruce, Nature Biotech., 2001, 19, 1168 CrossRef CAS; K. A. Thorn and K. R. Kennedy, Environ. Sci. Technol., 2002, 36, 3787 CrossRef CAS.
- C. J. McHugh, R. Keir, D. Graham and W. E. Smith, Chem. Commun., 2002, 580 RSC; R. Keir, E. Igata, M. Arundell, W. E. Smith, D. Graham, C. J. McHugh and J. M. Cooper, Anal. Chem., 2002, 74, 1503 CrossRef CAS.
- C. J. McHugh, W. E. Smith, R. Lacey and D. Graham, Chem. Commun., 2002, 2514 RSC.
- G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- A. N. Fitch, Nucl. Instrum. Methods Phys. Res., Sect. B, 1995, 97, 63 CrossRef CAS.
- K. Shankland, W. I. F. David and D. S. Sivia, J. Mater. Chem., 1997, 7, 569 RSC.
- A. Boultif and D. Louer, J. Appl. Crystallogr., 1991, 24, 987 CrossRef CAS.
- W. I. F. David, K. Shankland, J. Cole, S. Maginn, W. D. S. Motherwell, R. Taylor, DASH User Manual, Cambridge Crystallographic Data Centre, Cambridge, 2001 Search PubMed.
- W. I. F. David, K. Shankland and N. Shankland, Chem. Commun., 1998, 931 RSC; K. Shankland, L. McBride, W. I. F. David, N. Shankland and G. Steele, J. Appl. Cryst., 2002, 35, 443 CrossRef CAS.
- R. M. Vrcelj, J. N. Sherwood, A. R. Kennedy, H. G. Gallagher and T. Gelbrich, Cryst. Growth Des., 2003 DOI:10.1021/cg0340704, in press.
- For a detailed survey of nitro conformations in aromatic compounds see : A. Szumna, J. Jurczak and Z. Urbańczyk-Lipkowska, J. Mol. Struct., 2000, 526, 165 Search PubMed.
- A. Domenicano, Structural substituent effects in benzene derivatives in accurate molecular structures, eds. A. Domenicano and I. Hargittai, Oxford University Press, 1992, 437–468, ch. 18 Search PubMed.
- J. Maurin and T. M. Krygowski, J. Mol. Struct., 1988, 172, 413 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., 2002, B58, 380 CAS.
- K. Wozniak, H. He, J. Klinowski, W. Jones and E. Grech, J. Phys. Chem., 1994, 98, 13755 CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- R. S. Rowland and R. Taylor, J. Phys. Chem., 1996, 100, 7384 CrossRef CAS.
- B. Szczęsna and Z. Urbańczyk-Lipkowska, New J. Chem., 2002, 26, 243 RSC.
- S. L. Price, A. J. Stone, J. Lucas, R. S. Rowland and A. E. Thornley, J. Am. Chem. Soc., 1994, 116, 4910 CrossRef CAS.
- R. E. Dinnebier, M. Schweiger, B. Bildstein, K. Shankland, W. I. F. David, A. Jobst and S. van Smallen, J. Appl. Cryst., 2000, 33, 1199 CrossRef CAS.
- A. Huq and P. W. Stephens, J. Pharm. Sci., 2003, 92, 244 CrossRef CAS.
Footnote |
| † CCDC reference numbers 220762–220764. See http://www.rsc.org/suppdata/nj/b3/b309792g/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2004 |