Synthesis, structure and electrochemical properties of some oxime complexes of rhodium
Rama
Acharyya
a,
Falguni
Basuli
a,
Georgina
Rosair
b and
Samaresh
Bhattacharya
*a
aDepartment of Chemistry, Inorganic Chemistry Section, Jadavpur University, Kolkata, 700 032, India. E-mail: samaresh_b@hotmail.com
bSchool of Engineering & Physical Sciences – Chemistry, Heriot-Watt University, Edinburgh, EH14 4AS, UK
First published on 12th November 2003
Abstract
Reaction of the oximes of salicyladehyde (H2L1), 2-hydroxyacetophenone (H2L2) and 2-hydroxynaphthaldehyde (H2L3; general abbreviation H2L, where H2 stands for the two dissociable protons, one phenolic proton and one oxime proton) with [Rh(PPh3)3Cl] afforded a family of rhodium(III) complexes of the type [Rh(PPh3)2(HL)(L)]. The crystal structure of [Rh(PPh3)2(HL2)(L2)] has been determined by X-ray diffraction. One oxime ligand is coordinated via dissociation of only the phenolic proton, while the other oxime ligand is coordinated via dissociation of both the phenolic and oxime protons. Both the oxime ligands are coordinated as bidentate N,O-donors, forming six-membered chelate rings. The complexes are diamagnetic (low-spin d6, S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =0) and their 1H NMR spectra are in excellent agreement with their compositions. All three [Rh(PPh3)2(HL)(L)] complexes display intense absorptions in the visible and ultraviolet regions. Cyclic voltammetry on all the complexes shows two oxidations; the first one is observed within the range 0.61 to 0.76 V vs. SCE and the second one within 1.20 to 1.32 V vs. SCE. There is also one irreversible reduction between −1.05 and −1.30 V vs. SCE.
=0) and their 1H NMR spectra are in excellent agreement with their compositions. All three [Rh(PPh3)2(HL)(L)] complexes display intense absorptions in the visible and ultraviolet regions. Cyclic voltammetry on all the complexes shows two oxidations; the first one is observed within the range 0.61 to 0.76 V vs. SCE and the second one within 1.20 to 1.32 V vs. SCE. There is also one irreversible reduction between −1.05 and −1.30 V vs. SCE.
Introduction
The chemistry of rhodium complexes has been receiving considerable current attention,1 primarily because of the interesting reactivities exhibited by these complexes. As the properties of complexes are primarily dictated by the coordination environment around the metal center, complexation of rhodium by ligands of selected types has been of significant importance. The present work has originated from our interest in this area.2 As in our earlier works,2 Wilkinson's catalyst, viz. [Rh(PPh3)3Cl], has again been chosen as the rhodium starting material in the present study. This particular complex is known to undergo dissociation in solution, producing free PPh3 and a formally tricoordinated Rh(PPh3)2Cl species,3 which has been found to undergo simple complexation,2d as well as to promote interesting reactions such as cyclometallation via C–H activation,2b activation of molecular oxygen,2a reduction of hydroxamic acids2c and unusual chemical transformation of semicarbazones.2e Thus, Wilkinson's catalyst has also been found to serve as an useful synthon for the preparation of interesting complexes.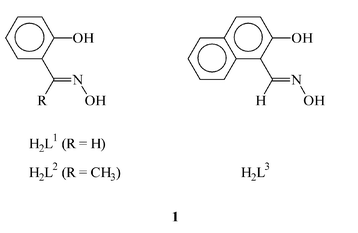
To interact with Wilkinson's catalyst, a group of three oxime ligands (1; viz. the oximes of salicylaldehyde, 2-hydroxyacetophenone and 2-hydroxynaphthaldehyde) containing a phenolic OH group, in addition to the oxime function, have been selected. These ligands are abbreviated in general as H2L, where H2 stands for the two dissociable protons, the phenolic proton and the oxime proton. These ligands are known to bind to metal ions, usually via dissociation of only the phenolic proton, as bidentate N,O-donors to form stable six-membered chelate rings.4 Under suitable experimental conditions, the oxime proton also dissociates.5 We have also observed these ligands to undergo the interesting oxime (>C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–OH) to imine (>CN–H) transformation upon reaction with [Ru(PPh3)3Cl2].6 It may be mentioned here that though rhodium complexes of different oxime ligands have received some attention,7 the chemistry of the rhodium complexes of these oxime ligands (1) appears to have remained unexplored. The primary intention of the present study has been to see how the oxime ligands (1) interact with Wilkinson's catalyst. Upon reaction of [Rh(PPh3)3Cl] with 1 a group of rhodium complexes of the type [Rh(PPh3)2(HL)(L)] has been obtained. The chemistry of these complexes is reported in this paper with special reference to their synthesis, structure and electrochemical properties.
N–OH) to imine (>CN–H) transformation upon reaction with [Ru(PPh3)3Cl2].6 It may be mentioned here that though rhodium complexes of different oxime ligands have received some attention,7 the chemistry of the rhodium complexes of these oxime ligands (1) appears to have remained unexplored. The primary intention of the present study has been to see how the oxime ligands (1) interact with Wilkinson's catalyst. Upon reaction of [Rh(PPh3)3Cl] with 1 a group of rhodium complexes of the type [Rh(PPh3)2(HL)(L)] has been obtained. The chemistry of these complexes is reported in this paper with special reference to their synthesis, structure and electrochemical properties.
Experimental
Materials and physical measurements
Rhodium trichloride was obtained from Arora Matthey (Kolkata, India) and triphenylphosphine was purchased from Loba Chemie (Mumbai, India). [Rh(PPh3)3Cl] was synthesized following a reported procedure.8 The oxime ligands were prepared by condensing the respective aldehyde or ketone with hydroxylamine. Purification of dichloromethane and acetonitrile, and preparation of tetrabutylammonium perchlorate (TBAP) for electrochemical work, were performed as before.9Microanalyses (C, H, N) were performed using a Heraeus Carlo Erba 1108 elemental analyzer. IR spectra were obtained on a Shimadzu FTIR-8300 spectrometer with samples prepared as KBr pellets. Electronic spectra were recorded on a JASCO V-570 spectrophotometer. Magnetic susceptibilities were measured using a PAR 155 vibrating sample magnetometer fitted with a Walker Scientific L75FBAL magnet. 1H NMR spectra were recorded in CDCl3 solution with a Bruker Avance DPX 300 NMR spectrometer using TMS as the internal standard. Electrochemical measurements were done in 1∶9 dichloromethane–acetonitrile solution (0.1 M TBAP) using a CH Instruments model 600A electrochemical analyzer. The addition of 10% dichloromethane was necessary to dissolve the complexes and acetonitrile was necessary for detection of the reductive response. A platinum disc working electrode, a platinum wire auxiliary electrode and an aqueous saturated calomel reference electrode (SCE) were used in a three-electrode configuration. Electrochemical measurements were made under a dinitrogen atmosphere. All electrochemical data were collected at 298 K and are uncorrected for junction potentials.
Synthetic procedures
=7.6 Hz, 2H), 5.84 (t, J=7.0 Hz, 2H), 6.01 (s, 2H), 6.51 (d, J=8.2 Hz, 2H), 6.64 (t, J=6.9 Hz, 2H), 6.9–7.5 (2PPh3), 19.38 (s, 1H). UV-vis (CH2Cl2): λmax/nm (ε/M−1 cm−1) 44210
(6200), 39810
(8200), 29610
(26800).
300).
=7.8 Hz, 2H), 6.99 (d, J=9.0 Hz, 2H), 7.32 (d, J=8.3 Hz, 2H), 7.33 (s, 2H), 7.40 (d, J=6.9 Hz, 2H), 7.41–7.58 (4H), 7.0–7.8 (2PPh3), 19.41 (s, 1H). UV-vis (CH2Cl2): λmax/nm (ε/M−1 cm−1) 46810
(6900), 432 (9700), 33210
(17250).
X-Ray crystallography
Single crystals of [Rh(PPh3)2(HL2)(L2)] were grown by slow evaporation of an acetonitrile solution of the complex. Selected crystal data and data collection parameters are given in Table 1. Data were collected on a Bruker P4 diffractometer using graphite-monochromated MoKα radiation (λ=0.71069 Å) by ϕ and ω scans. The data were corrected for empirical absorption on the basis of psi scans. X-Ray structure solution and refinement were done using the SHELXS-97 and SHELXL-97 programs.11 Hydrogen atoms were fixed at calculated positions and were refined using a riding mode. The structure was solved by direct methods.†
| Formula | C52H45N2O4P2Rh |
| Formula weight | 926.75 |
| Crystal system | Monoclinic |
| Space group | P21/n |
| a/Å | 11.168(3) |
| b/Å | 17.158(2) |
| c/Å | 23.129(6) |
| β/deg | 96.68(2) |
| U/Å3 | 4401.9(17) |
| Z | 4 |
| T/K | 100(2) |
| μ/mm−1 | 0.509 |
| Reflections collected | 9565 |
| Independent reflections | 7654 |
| R int | 0.0908 |
| R 1 [I > 2σ(I)] | 0.0578 |
| wR 2 [I > 2σ(I)] | 0.1113 |
Results and discussion
Reaction of the oxime ligands (H2L, 1) with [Rh(PPh3)3Cl] proceeds smoothly in refluxing benzene in the presence of triethylamine to afford the [Rh(PPh3)2(HL)(L)] complexes in decent yields. Triethylamine has been used as a base to favor the dissociation of the acidic protons. Elemental (C, H, N) analytical data of the complexes are consistent with their compositions. The net synthetic reaction can be expressed as in eqn. 1:| [RhI(PPh3)3Cl]+2H2L→[RhIII(PPh3)2(HL)(L)]+PPh3+HCl+2H++2e− | (1) |
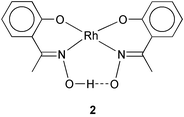
As the oxime ligands 1 are unsymmetric bidentate in nature, these [Rh(PPh3)2(HL)(L)] complexes may exist, in principle, in several geometric isomeric forms. To determine the stereochemistry of these complexes, the structure of one representative member of this family, viz. [Rh(PPh3)2(HL2)(L2)], has been determined by X-ray crystallography. A view of the complex molecule is shown in Fig. 1 and selected bond parameters are listed in Table 2. Both the partly deprotonated oxime ligand (HL2) and the fully deprotonated oxime ligand (L2) are coordinated to rhodium as bidentate N,O-donors, forming six-membered chelate rings (2) with bite angles of ∼88°. The two coordinated oxime ligands constitute the pseudo-equatorial plane with the metal at the center, with the phenolate oxygens being cis as are the oxime nitrogens (2). The coordinated oxime ligands are intramolecularly hydrogen-bonded (2) through the oxime hydrogen (in HL2) and the oximato oxygen (in L2). The two triphenylphosphines have taken up the remaining two axial positions and hence they are mutually trans. The N2O2P2Rh core is distorted from ideal octahedral geometry, as reflected in all the bond parameters around rhodium. The RhN2O2 fragment is almost planar, however, the phenyl rings of the oxime ligands do not lie in the same plane as the RhN2O2 fragment, primarily due to the sp3 character of the phenolate oxygens. The oxime fragments also do not lie in the same plane as the corresponding phenyl rings, due to a rotation of these fragments about the C(phenyl)–C(oxime) bonds. The Rh–N, Rh–O and Rh–P lengths are quite normal and so are the phenolic C–O lengths and the C–N and N–O distances in the oxime fragment.2,7i,7m,7n,7u,7v As all three complexes have been synthesized similarly and display similar properties (vide infra), the other two [Rh(PPh3)2(HL)(L)] complexes are assumed to have the same structure as [Rh(PPh3)2(HL2)(L2)].
| Rh(1)–N(1) | 2.038(5) | N(1)–Rh(1)–O(2) | 176.50(18) |
| Rh(1)–N(2) | 2.032(5) | N(2)–Rh(1)–O(1) | 177.98(18) |
| Rh(1)–O(1) | 2.049(4) | P(1)–Rh(1)–P(2) | 175.78(6) |
| Rh(1)–O(2) | 2.026(4) | N(1)–Rh(1)–O(1) | 87.37(17) |
| Rh(1)–P(1) | 2.3953(17) | N(2)–Rh(1)–O(2) | 88.88(17) |
| Rh(1)–P(2) | 2.3917(17) | N(1)–Rh(1)–N(2) | 94.62(19) |
| C(17)–N(1) | 1.306(7) | O(1)–Rh(1)–O(2) | 89.14(15) |
| N(1)–O(11) | 1.372(6) | Rh(1)–N(1)–O(11) | 115.2(3) |
| C(27)–N(2) | 1.296(7) | Rh(1)–N(2)–O(22) | 115.2(3) |
| N(2)–O(22) | 1.373(6) | ||
| C(11)–O(1) | 1.335(7) | ||
| C(21)–O(2) | 1.327(7) |
![View of the [Rh(PPh3)2(HL2)(L2)] molecule.](/image/article/2004/NJ/b309412j/b309412j-f1.gif) | ||
| Fig. 1 View of the [Rh(PPh3)2(HL2)(L2)] molecule. | ||
Magnetic susceptibility measurements show that the [Rh(PPh3)2(HL)(L)] complexes are diamagnetic, which is in accordance with the trivalent state of rhodium (low-spin d6, S=0) in these complexes. 1H NMR spectra of all the complexes show broad signals for the PPh3 protons within the 6.9–7.8 ppm range. Signals for the coordinated oxime ligands are also observed in the expected regions. In the spectrum of the [Rh(PPh3)2(HL1)(L1)] complex, a distinct signal has been observed at 19.38 ppm, which is attributable to the intramolecularly hydrogen-bonded oxime hydrogen. Besides the broad signals for the PPh3 ligands, five (one singlet, two doublets and two triplets) distinct signals (2H each) have been observed in the aromatic region due to the coordinated oxime ligands (HL1 and L1). The singlet is assignable to the azomethine proton, while the doublets and triplets are due to the four phenyl protons of each oxime ligand. In the [Rh(PPh3)2(HL2)(L2)] complex, a single methyl signal is observed at 1.13 ppm and the signal for the intramolecularly hydrogen-bonded oxime hydrogen is observed at 19.32 ppm. The other expected signals from the coordinated oxime ligands have been observed in the 6.06–6.82 ppm region. In the [Rh(PPh3)2(HL3)(L3)] complex, the signal for the intramolecularly hydrogen-bonded oxime hydrogen is observed at 19.41 ppm, the azomethine proton signal is at 7.33 ppm and the aromatic proton signals of the coordinated oxime ligands lie within 6.89–7.58 ppm. The 1H NMR spectral data thus indicate that the [Rh(PPh3)2(HL)(L)] complexes have a pseudo-C2 symmetry in solution and are therefore consistent with the composition and stereochemistry of the complexes.
Infrared spectra of the [Rh(PPh3)2(HL)(L)] complexes show three strong bands near 525, 695 and 745 cm−1, which are attributable to the Rh(PPh3)2 fragment.2 Comparison with the spectrum of [Rh(PPh3)3Cl] shows the presence of some additional bands in the spectra of the [Rh(PPh3)2(HL)(L)] complexes (e.g., near 1597, 1331, 1306 and 918 cm−1), which must be due to the coordinated oxime ligands. Electronic spectra of the [Rh(PPh3)2(HL)(L)] complexes have been recorded in dichloromethane solution. Each complex shows several intense absorptions in the visible and ultraviolet region. The absorptions in the ultraviolet region are assignable to transitions within the ligand orbitals and those in the visible region are probably due to charge-transfer transitions involving both metal and ligand orbitals.
For a better understanding of the nature of the transitions in the visible region, qualitative EHMO calculations have been performed12 on computer-generated models of all three complex molecules in which phenyl rings of the PPh3 ligands have been replaced by hydrogens. A partial MO diagram is displayed in Fig. 2 and composition of some selected molecular orbitals is given in Table 3. The highest occupied molecular orbital (HOMO) and the next two filled orbitals (HOMO−1 and HOMO−2) have major contribution from the dxy, dyz and dzx orbitals of rhodium13 and hence these three filled orbitals may be regarded as components of the metal t2 orbitals. The lowest unoccupied molecular orbital (LUMO) and the next couple of vacant orbitals (e.g., LUMO+1 and LUMO+2) are localized almost entirely on different parts of the oxime ligands. Hence the absorptions in the visible region may be assigned to charge-transfer transitions occurring from the filled t2 orbitals of rhodium to the vacant π* orbitals of the oxime ligands.
| Compound | Contributing fragments | % Contribution of fragments to | |||||
|---|---|---|---|---|---|---|---|
| HOMO | HOMO−1 |
HOMO−2 |
LUMO | LUMO+1 |
LUMO+2 |
||
| [Rh(PPh3)2(HL1)(L1)] | Rh | 59 | 64 | 81 | 4 | 3 | – |
| HL1 | 12 | 17 | 9 | 92 | 0 | 99 | |
| L1 | 23 | 12 | 4 | 0 | 94 | 0 | |
| [Rh(PPh3)2(HL2)(L2)] | Rh | 57 | 62 | 82 | 3 | 3 | – |
| HL2 | 12 | 19 | 9 | 95 | 0 | 99 | |
| L2 | 24 | 12 | 4 | 0 | 91 | 0 | |
| [Rh(PPh3)2(HL3)(L3)] | Rh | 52 | 58 | 46 | 1 | – | – |
| HL3 | 18 | 21 | 16 | 95 | 0 | 88 | |
| L3 | 20 | 14 | 28 | 0 | 94 | 4 | |
![Partial molecular orbital diagram of [Rh(PPh3)2(HL2)(L2)].](/image/article/2004/NJ/b309412j/b309412j-f2.gif) | ||
| Fig. 2 Partial molecular orbital diagram of [Rh(PPh3)2(HL2)(L2)]. | ||
Electrochemical properties of the [Rh(PPh3)2(HL)(L)] complexes have been studied by cyclic voltammetry. Voltammetric data are presented in Table 4 and a selected voltammogram is displayed in Fig. 3. Each complex shows two oxidative responses (OxI and OxII) on the positive side of the SCE and one reductive response (RedI) on the negative side. The first oxidative response is observed within 0.61–0.76 V and is assigned to rhodium(III)-to-rhodium(IV) oxidation. This oxidation is quasi-reversible in nature with a peak-to-peak separation (ΔEp) of 66–80 mV; the anodic peak current (ipa) is greater than the cathodic peak current (ipc). The one-electron nature of this oxidation has been established by comparing its current height (ipa) with that of the ferrocene/ferrocenium couple under identical experimental conditions. The second oxidative response (observed within 1.20–1.32 V) is irreversible in nature and this oxidation is believed to be centered on the oxime ligand. The reductive response (observed below −1.0 V) is also irreversible and in view of the composition of the LUMO (vide supra) it is assigned to reduction of the coordinated oxime ligand.
| Compound | E vs. SCE/V | ||
|---|---|---|---|
| OxIIb | OxIc | RedId | |
| a In 1∶9 dichloromethane–acetonoitrile; supporting electrolyte TBAP; scan rate 50 m s−1. b E pa value. c E 1/2 value (ΔEp value). d E pc value. | |||
| [Rh(PPh3)2(HL1)(L1)] | 1.32 | 0.76(80) | −1.05 |
| [Rh(PPh3)2(HL2)(L2)] | 1.25 | 0.75(70) | −1.30 |
| [Rh(PPh3)2(HL3)(L3)] | 1.20 | 0.61(66) | −1.30 |
![Cyclic voltammogram of [Rh(PPh3)2(HL2)(L2)] in 1∶9 dichloromethane–acetonitrile solution (0.1 M TBAP) at a scan rate of 50 mV s−1.](/image/article/2004/NJ/b309412j/b309412j-f3.gif) | ||
| Fig. 3 Cyclic voltammogram of [Rh(PPh3)2(HL2)(L2)] in 1∶9 dichloromethane–acetonitrile solution (0.1 M TBAP) at a scan rate of 50 mV s−1. | ||
Conclusion
The present study shows that the oxime ligands (H2L, 1) can bind smoothly to rhodium upon reaction with [Rh(PPh3)3Cl], affording complexes of the type [Rh(PPh3)2(HL)(L)], where the coordinated oxime ligands are intramolecularly hydrogen-bonded (2). The disposition of the oxime oxygens in these [Rh(PPh3)2(HL)(L)] complexes points to their possible coordination to another metal ion, via dissociation of the intramolecularly hydrogen-bonded oxime hydrogen, leading to the formation of polymetallic assemblies. Such a possibility is currently under exploration.Acknowledgements
Financial assistance received from Council of Scientific and Industrial Research, New Delhi [Grant No. 01(1675)/00/EMR-II] is gratefully acknowledged. The authors thank Professor Saswati Lahiri of the Department of Organic Chemistry, Indian Association for the Cultivation of Science, Kolkata, for her help.References
- (a) C. E. Housecroft, Coord. Chem. Rev., 1992, 115, 191 CrossRef CAS; (b) C. E. Housecroft, Coord. Chem. Rev., 1994, 134, 307 CAS; (c) C. E. Housecroft, Coord. Chem. Rev., 1995, 146, 235 CrossRef CAS; (d) M. J. Hannon, Coord. Chem. Rev., 1997, 162, 477 CrossRef CAS; (e) A. M. Trezeciak and J. J. Ziolkowski, Coord. Chem. Rev., 1999, 190–192, 883 CrossRef CAS.
- (a) S. Dutta, S.-M. Peng and S. Bhattacharya, Inorg. Chem., 2000, 39, 2231 CrossRef CAS; (b) S. Dutta, S.-M. Peng and S. Bhattacharya, J. Chem. Soc., Dalton Trans., 2000, 4623 RSC; (c) A. Das, F. Basuli, S.-M. Peng and S. Bhattacharya, Inorg. Chem., 2002, 41, 440 CrossRef CAS; (d) S. Dutta, F. Basuli, S.-M. Peng, G.-H. Lee and S. Bhattacharya, New J. Chem., 2002, 26, 1607 RSC; (e) I. Pal, S. Dutta, F. Basuli, S. Goverdhan, S.-M. Peng, G.-H. Lee and S. Bhattacharya, Inorg. Chem., 2003, 42, 4338 CrossRef CAS.
- F. A. Cotton and G. Wilkinson, Advanced Inorganic Chemistry, Wiley Eastern, New Delhi, 1985, p. 788 Search PubMed.
- (a) A. Chakravorty, Coord. Chem. Rev., 1974, 13, 1 CrossRef CAS; (b) V. Y. Kukushkin and A. J. L. Pomberio, Coord. Chem. Rev., 1999, 181, 147 CrossRef CAS.
- (a) P. Chaudhuri, M. Hess, T. Weyhermuller, E. Bill, H.-J. Haupt and U. Florke, Inorg. Chem. Commun., 1998, 1, 39 CrossRef CAS; (b) C. N. Verani, E. Bothe, D. Burdinski, T. Weyhermuller, U. Florke and P. Chaudhuri, Eur. J. Inorg. Chem., 2001, 2161 CrossRef CAS.
- A. K. Das, S.-M. Peng and S. Bhattacharya, J. Chem. Soc., Dalton Trans., 2000, 181 RSC.
- (a) K. C. Kalia and A. Chakravorty, Inorg. Chem., 1969, 8, 2586 CrossRef CAS; (b) H. Onoue and I. Moritani, J. Organomet. Chem., 1972, 44, 189 CAS; (c) J. P. Collman, D. W. Murphy and G. Dolcetti, J. Am. Chem. Soc., 1973, 95, 2687 CrossRef CAS; (d) M. Nanoyama, Inorg. Nucl. Chem. Lett., 1975, 11, 123 Search PubMed; (e) H. Yukimasa, H. Sawai and T. Takizawa, Makromol. Chem., 1980, 1, 579 CrossRef CAS; (f) C. A. Johnson and A. J. Nielson, Polyhedron, 1982, 1, 501 CrossRef CAS; (g) S. Siripaisarnpipat and E. O. Schlemper, Inorg. Chem., 1983, 22, 282 CrossRef CAS; (h) H. Brunner and R. Becker, Angew. Chem., 1984, 96, 221 CAS; (i) S. Siripaisarnpipat and E. O. Schlemper, Inorg. Chem., 1984, 23, 330 CrossRef; (j) R. D. Garlatti, G. Tauzher, M. Blaschich and G. Costa, Inorg. Chim. Acta, 1985, 105, 129 CrossRef; (k) J. Charalambous, K. Henrick, Y. Musa, R. G. Rees and R. N. Whiteley, Polyhedron, 1987, 6, 1509 CrossRef CAS; (l) T. Lynde-Kernell and E. O. Schlemper, J. Coord. Chem., 1988, 16, 347 CAS; (m) G. E. Efe and E. O. Schlemper, Polyhedron, 1991, 10, 1611 CrossRef CAS; (n) G. E. Efe, M. R. A. Pillani, E. O. Schlemper and D. E. Troutner, Polyhedron, 1991, 10, 1617 CrossRef CAS; (o) G. E. Efe and E. O. Schlemper, Polyhedron, 1992, 11, 2447 CrossRef CAS; (p) G. E. Efe and E. O. Schlemper, Polyhedron, 1993, 12, 2869 CrossRef CAS; (q) R. Dreos, G. Tauzher, S. Geremia, L. Randaccio, F. Asaro, G. Pellizer, C. Tavagnacco and G. Costa, Inorg. Chem., 1994, 33, 5404 CrossRef CAS; (r) S. Ganguly, V. Manivannam and A. Chakravorty, J. Chem. Soc., Dalton Trans., 1998, 461 RSC; (s) D. Steinborn, M. Rausch and C. Bruhn, J. Organomet. Chem., 1998, 561, 191 CrossRef CAS; (t) G. Costa, C. Tavagnacco and R. Mahajan, Bull. Electrochem., 1998, 14, 78 Search PubMed; (u) V. Y. Kukushkin, I. V. Ilichev, G. Wagner, F. D. Silva, J. R. Joao and A. J. L. Pomberio, J. Chem. Soc., Dalton Trans., 1999, 3047 RSC; (v) X.-X. Liu and W.-T. Wong, Eur. J. Inorg. Chem., 2001, 511 CrossRef; (w) R. Dreos, G. Nardin, L. Randaccio, P. Siega and G. Tauzher, Eur. J. Inorg. Chem., 2002, 2885 CrossRef CAS.
- J. A. Osborn and G. Wilkinson, Inorg. Synth., 1967, 10, 67 CAS.
- (a) D. T. Sawyer and J. L. Roberts, Jr., Experimental Electrochemistry for Chemists, Wiley, New York, 1974, pp. 167–215 Search PubMed; (b) M. Walter and L. Ramaley, Anal. Chem., 1973, 45, 165 CAS.
- This absorption appears as a shoulder.
- G. M. Sheldrick, SHELXS-97, Program for solution of crystal structures, University of Göttingen, Germany, 1997 Search PubMed; G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- (a) C. Mealli and D. M. Proserpio, CACAO Version 4.0, Firenze, Italy, July 1994 Search PubMed; (b) C. Mealli and D. M. Proserpio, J. Chem. Educ., 1990, 67, 399 CAS.
- The HOMO-2 in [Rh(PPh3)2(HL3)(L3)Cl] has slightly less metal character.
Footnote |
| † CCDC reference number 220837. See http://www.rsc.org/suppdata/nj/b3/b309412j/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2004 |