A fluorescence study of (4-{(1E,3E)-4-[4-(dimethylamino)phenyl]buta-1,3-dienyl}phenyl)methanol and its bioconjugates with bovine and human serum albumins
Anil K.
Singh
* and
Manjula
Darshi
Department of Chemistry, Indian Institute of Technology, Bombay, Powai, Mumbai, 400 076, India. E-mail: retinal@chem.iitb.ac.in; Fax: +91 22 2576 7152
First published on 12th November 2003
Abstract
Electronic absorption and fluorescence properties of (4-{(1E,3E)-4-[4-(dimethylamino)phenyl]buta-1,3-dienyl}phenyl)methanol in different media, including organic solvents of varying polarity at 298 K, an ethanol–methanol matrix at 77 K and protein environments, have been examined. The diene shows solvatochromic fluorescence, which is correlated with empirical solvent polarity parameters. The red-shifted fluorescence emission of the diene is attributed to a polar, conformationally relaxed intramolecular charge transfer excited state. Further, the diene has been covalently attached to bovine serum albumin and human serum albumin and the fluorescence probe properties of the protein-diene conjugates have been examined in phosphate buffer.
Introduction
The strategy of studying the stereoelectronic nature of biomolecular binding sites by critically examining spectroscopic changes in a probe when bound to the binding site is well-known.1–9 Fluorescence spectroscopy and probe molecules that exhibit solvatochromic fluorescence have been particularly useful for this purpose.10–15 Since the intensity and the maximum wavelength of the fluorescence emission also depends on solvent polarity, compounds exhibiting solvatochromic dual fluorescence emission can be used as probes of environmental polarity. We have recently reported that certain diphenylbutadienes are capable of solvatochromic fluorescence emission and that these can be used as fluorescence probes for studying the microenvironment of organised assemblies.16–20 The large red shift in the fluorescence emission of such compounds bearing strong donor or acceptor groups on the phenyl ring has been attributed to conformational relaxation of the excited state and intramolecular charge transfer.Herein we report a new fluorescence probe, namely (4-{(1E,3E)-4-[4-(dimethylamino)phenyl]buta-1,3-dienyl}phenyl)methanol (6) that can be used to study the microenvironment of proteins. The fluorescence probe properties of 6 are based on its solvent polarity dependence and novel substituent-tuned fluorescence emission behaviour. Its excited state is characterised by charge transfer process. It shows large Stokes' shifts and its fluorescence quantum yield (Φf) is greater than that of other known probe dienes (e.g., the nitro diphenyldienes),18 particularly in biologically significant aqueous medium. To examine the usefulness of this diene as a fluorescence probe for proteins, it has been covalently attached to bovine serum albumin (BSA) and human serum albumin (HSA) and the fluorescence characteristics of the resulting protein conjugates 8 and 9 have been investigated.
Experimental
Materials and general procedures
Starting materials for the synthesis, BSA and quinine sulfate, were procured from local suppliers and used as received. Fatty-acid-free HSA was obtained from Sigma Chemical Company (USA). Diphosgene was procured from IICT (Hyderabad, India). Sephadex-G50 used for gel filtrations was from Pharmacia Biotechnology (Sweden). Solvents used for the synthesis of dienes and for photophysical studies were from Spectrochem India Ltd. (Mumbai) and were freshly dried and distilled prior to use. Thin layer chromatography (TLC) and column chromatography were run on silica gel and neutral alumina, respectively. Deionised and double-distilled water was used for the preparation of all aqueous solutions. Phosphate buffer used was 0.1 M (pH 7.4).Melting points were determined on Veego melting point apparatus by the capillary method and are uncorrected. UV-vis measurements were carried out on a Shimadzu UV-160 spectrophotometer. IR spectra in KBr discs were recorded on an Impact 400 Nicolet FTIR spectrophotometer. 1H-NMR (300 MHz) spectra in CDCl3 as solvent and TMS as internal standard were measured on a Varian VXR 300 FT NMR spectrometer. Elemental analyses were done on a Thermoquest CE instrument-1112 series CHNS autoanalyser. Steady state fluorescence measurements were carried out on a DM1B microprocessor controlled Spex-112 Fluorolog spectrofluorimeter with slit widths of 1 and 1.5 mm for the excitation and emission monochromators, respectively. The low temperature fluorescence studies were done using an ethanol–methanol (1∶1, v/v) glass at 77 K using a Spex-112 Fluorolog equipped with Spex-1932 F accessories. The fluorescence quantum yields (Φf) at ambient temperature were determined using quinine sulfate as the standard21
(Φf![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =0.55, the revised value is 0.53).22 The low temperature Φf are relative to the quinine sulfate Φf at ambient temperature.
=0.55, the revised value is 0.53).22 The low temperature Φf are relative to the quinine sulfate Φf at ambient temperature.
Syntheses
The synthetic routes for diene 6 and protein conjugates 8 and 9 are outlined in Scheme 1 and the details are described below.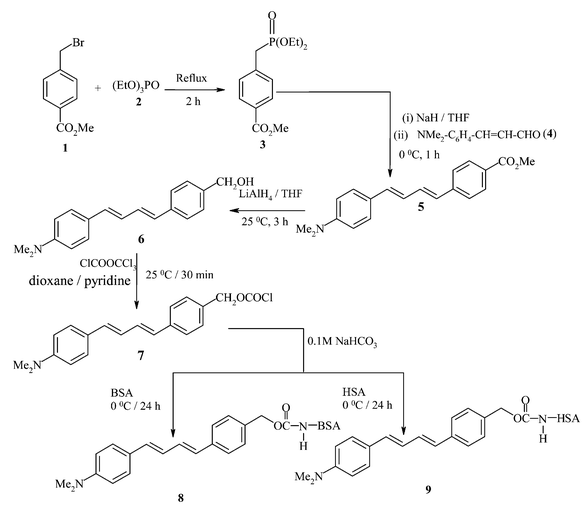 | ||
| Scheme 1 | ||
°C under N2 atmosphere. 4-N,N-Dimethylamino cinnamaldehyde (4) taken up in dry THF was added dropwise to the stirred reaction mixture. The reaction mixture was slowly brought to room temperature and the stirring was allowed to continue until most of the aldehyde had disappeared, as indicated by TLC in 10% ethyl acetate in petroleum ether (60–80°C). After completion of the reaction, the reaction mixture was quenched with brine and taken up in diethyl ether. The ether layer was washed with water and dried over anhydrous sodium sulfate. Removal of ether on a rotary evaporator yielded solid material, which was purified by column chromatography using 20% ethyl acetate in petroleum ether (60–80°C) as eluent; methyl 4-{(1E,3E)-4-[4-(dimethylamino)phenyl]buta-1,3-dienyl}benzoate (5) was obtained in 62% yield. M.p. 209–210°C. UV-vis (MeOH)
λmax/nm: 395 (ε 26644 mol−1 cm−1 l). IR νmax/cm−1: 1726 (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O str), 1606 (–CC str), 1368 (–C–N str of –Ar–N), 958 (–C–H def of trans alkene). 1H NMR (CDCl3, 300 MHz)
δ: 2.99 (6H, s, NMe2), 3.91 (3H, s, –COOMe), 6.58 (1H, d, J=15.48, NMe2–C6H4–CHCH–), 6.67 (1H, d, J=15.56 Hz, MeOOC–C6H4–CHCH–), 6.69 (2H, d, AB coupling J=8.79 Hz, NMe2–C6H4–), 6.79 (1H, dd, J=15.65, 10.05 Hz, Me2N–C6H4–CHCH–CH), 7.05 (1H, dd, J=15.36, 9.92 Hz, MeOOC–C6H4–CHCH–), 7.35 (2H, d, AB coupling J=8.79 Hz Me2N–C6H4–), 7.45 (2H, d of AB coupling, J=8.24 Hz, MeOOC–C6H4–), 7.98 (2H, dd of aromatic quartet, J=8.24, 1.83 Hz, MeOOC–C6H4–). Anal. calcd for C20H21NO2
(307.40): C, 78.15; H, 6.89; N, 4.56; found: C, 78.19; H, 6.74; N, 4.63.
O str), 1606 (–CC str), 1368 (–C–N str of –Ar–N), 958 (–C–H def of trans alkene). 1H NMR (CDCl3, 300 MHz)
δ: 2.99 (6H, s, NMe2), 3.91 (3H, s, –COOMe), 6.58 (1H, d, J=15.48, NMe2–C6H4–CHCH–), 6.67 (1H, d, J=15.56 Hz, MeOOC–C6H4–CHCH–), 6.69 (2H, d, AB coupling J=8.79 Hz, NMe2–C6H4–), 6.79 (1H, dd, J=15.65, 10.05 Hz, Me2N–C6H4–CHCH–CH), 7.05 (1H, dd, J=15.36, 9.92 Hz, MeOOC–C6H4–CHCH–), 7.35 (2H, d, AB coupling J=8.79 Hz Me2N–C6H4–), 7.45 (2H, d of AB coupling, J=8.24 Hz, MeOOC–C6H4–), 7.98 (2H, dd of aromatic quartet, J=8.24, 1.83 Hz, MeOOC–C6H4–). Anal. calcd for C20H21NO2
(307.40): C, 78.15; H, 6.89; N, 4.56; found: C, 78.19; H, 6.74; N, 4.63.
The ester 5 prepared as above was converted to the corresponding alcohol 6 by reducing it with lithium aluminium hydride. In a typical reaction, lithium aluminium hydride (0.036 g, 9.6 mM) was added into 10 mL dry THF under nitrogen atmosphere and stirred for 10 min. To this was added ester 5
(0.1 g, 3.2 mM) taken up in 10 mL dry THF and the reaction mixture was stirred at room temperature for 3 h. The progress of the reaction was monitored by TLC in 40% ethyl acetate in petroleum ether (60–80°C) and, after completion of the reaction, the reaction mixture was cooled to 0°C and was slowly neutralised with 50 mL saturated ammonium chloride solution. The quenched reaction mixture was extracted with ethyl acetate and the organic layer was dried over anhydrous sodium sulfate. Evaporation of ethyl acetate under reduced pressure yielded a yellow solid, which was further purified by column chromatography using 30% ethyl acetate in petroleum ether (60–80°C) as eluent; (4-{(1E,3E)-4-[4-(dimethylamino)phenyl]buta-1,3-dienyl}phenyl)methanol (6) was obtained in 67% yield. M.p. 215–218°C. UV-vis (1,4-dioxane)
λmax/nm: 372 (ε 40300 mol−1 cm−1 l). IR (KBr) νmax/cm−1: 3400–3500 (–OH), 1599 (–CC–), 1365 (–C–N str of –Ar–N). 1H NMR (CDCl3, 300 MHz)
δ: 2.98 (6H, s, NMe2), 4.67 (2H, d, –CH2OH) 7.41 (2H, d, AB coupling, J=8.24, 2.01 Hz, HOH2C–C6H4–), 7.31 (2H, d, AB coupling, J=8.24, 2.01 Hz, HOH2C–C6H4–), 7.35 (2H, d, AB coupling, J=8.97, 2.01 Hz, Me2N–C6H4–), 6.69 (2H, d, AB coupling, J=8.97, 2.01 Hz, NMe2–C6H4–), 6.95 (1H, dd, J=15.19, 8.08 Hz, HOH2C–C6H4–CHCH–), 6.76 (1H, dd, J=15.19, 8.05 Hz, Me2N–C6H4–CHCH–), 6.60 (1H, d, J=15.19 Hz, HOH2C–C6H4–CH–), 6.56 (1H, d, J=15.19 Hz, Me2N–C6H4–CH–). Anal. calcd for C19H21NO (279.39): C, 81.68; H, 7.58; N, 5.01; found: C, 81.74; H, 7.51; N, 5.78.
Aliquots of an ethanolic solution of the chloride 7 (20–80 µL) were added to 2 mg of the proteins taken up in 1 mL aq. NaHCO3
(0.1 M) solution. The mixture was kept overnight at 4°C. After this, the reaction mixture was passed through a Sephadex-G50 column to remove unreacted diene. The protein-diene conjugates were further dialysed for 6 h against 1 L of phosphate buffer at 4°C to ensure total removal of unreacted diene. The mixture was diluted to 5 mL with phosphate buffer and then its UV-vis absorption spectrum was measured. The average number of diene residues bound to the protein was estimated by known procedures from the concentration of the protein and the concentration of the diene in the conjugates using the following equation:24D/P=Cd/Cp where Cd=Aλmax/εd, Cp=[A280−(AλmaxF)]/εp, and F=Ad(280)/Ad. In this equation D/P is the number of chromophores bound to the protein, Cd is the concentration of chromophore in the conjugate (mol L−1), Cp is the concentration of protein in the conjugate (mol L−1), Aλmax is the absorbance of chromophore in the conjugate at λmax
(=325 nm), εd is the absorption coefficient due to diene chromophore at 325 nm, Ad(280) is absorbance of free chromophore at 280 nm, Ad is the absorbance of diene chromophore at 325 nm, A280 is the absorbance of protein in conjugate at 280 nm, and εp is the extinction coefficient of protein at 280 nm.
Five conjugates (8, 8a–8d, and 9, 9a–9d) of each protein having a different number of diene units were prepared. The number of diene units bound to the proteins depended on the aliquot of diene solution added to the protein during the coupling reaction. Conjugate 8 and 9 were found to contain a maximum of 4.6 and 4.25 units of the diene chromophore. The other protein conjugates were found to contain the following concentrations of the diene units: 8a, 1.9×10−5 M (D/P
∼0.99); 8b, 2.7×10−5 M (D/P
∼1.42); 8c, 5.0×10−5 M (D/P
∼2.7); 8d, 7.7×10−5 M (D/P
∼4.1); 9a, 2.1×10−5 M (D/P
∼1.27); 9b, 2.8×10−5 M (D/P
∼1.7); 9c, 5.0×10−5 M (D/P
∼03.03); 9d, 6.4×10−5 M (D/P
∼3.88).
Results and discussion
Electronic absorption and fluorescence properties of 6 in organic solvents and 1,4-dioxane–water mixtures
The UV-vis absorption and fluorescence spectral data of diene 6 in homogeneous media (organic solvents) and in 1,4-dioxane–water mixtures are summarised in Tables 1 and 2. For comparison purposes the absorption and fluorescence spectral data of the parent diene N,N-dimethyl-N-[4-phenylbuta-(1E,3E)-dienyl]phenylamine in organic solvents are also given in Table 1, while the data in 1,4-dioxane–water mixtures was already reported.20 Similar to the parent diene, no significant changes are observed in the absorption and fluorescence excitation spectra of 6 in the various solvents examined, except that in aprotic polar solvents the absorption maximum is slightly red-shifted while in protic solvents like methanol and water it experiences a blue shift. In 1,4-dioxane–water mixtures a similar trend is observed, that is the absorption maximum tends to decrease as the water content is increased. However, the fluorescence excitation wavelength does not change. These absorption spectral features can be due to ground state hydrogen bond interactions and aggregate formation, particularly in water in which the diene is not freely soluble.| Solvent | λ abs max/nm | λ f max/nm | λ ex max/nm | Stokes' shift/cm−1 | Φ f |
|---|---|---|---|---|---|
| Heptane | 367 (364) | 430, 457sh (424) | 366 (366) | 3992.(3887) | 0.167 (0.125) |
| 1,4-Dioxane | 372 (368) | 441sh, 458 (440) | 369 (367) | 4205 (4446) | 0.065 (0.055) |
| THF | 374 (369) | 446sh, 477 (474) | 370 (372) | 5995 (6003) | 0.059 (0.052) |
| Acetonitrile | 369 (371) | 491 (492) | 367 (362) | 6733 (6628) | 0.046 (0.028) |
| Methanol | 354 (370) | 491 (484) | 368 (369) | 7881 (6365) | 0.031 (0.029) |
| DMF | 376 (378) | 493 (496) | 370 (376) | 6311 (6293) | 0.037 (0.045) |
| Water | 310 | 521 | 365 | 12252 |
0.009 |
| % Water in 1,4-dioxane | Dielectric constant10ε | λ abs max/nm | λ f max/nm | λ ex max/nm | Stokes' shift/cm−1 | Φ f |
|---|---|---|---|---|---|---|
| 0 | 2 | 372 | 458 | 369 | 4205 | 0.065 |
| 10 | 5 | 371 | 478 | 369 | 6033 | 0.051 |
| 20 | 10 | 372 | 486 | 370 | 6305 | 0.047 |
| 30 | 16 | 370 | 491 | 370 | 6660 | 0.041 |
| 40 | 25 | 368 | 496 | 370 | 7012 | 0.038 |
| 50 | 32 | 365 | 499 | 368 | 7357 | 0.034 |
| 60 | 42 | 368 | 503 | 370 | 7293 | 0.033 |
| 70 | 50 | 364 | 504 | 370 | 7631 | 0.030 |
In contrast to a very moderate solvent polarity effect on the absorption maximum and almost no effect on the fluorescence excitation spectrum, there is a marked influence of the solvent polarity on the fluorescence emission behaviour of the diene (Fig. 1). The fluorescence emission gets red-shifted by 91 nm when the diene is in a polar protic medium like water as compared to that in a non-polar solvent like heptane. In the relatively non-polar media of heptane, 1,4-dioxane and THF, the diene shows two fluorescence bands centred at around 430 and 470 nm. With increase in solvent polarity the peak at 430 nm disappears and there is red shift in the longer wavelength peak. Even though in water there is large blue shift in the absorption maximum, the fluorescence emission maximum is highly red-shifted. The fluorescence excitation maximum in organic solvents is similar to the absorption maximum in the same solvents. However, in water the excitation maximum is considerably red-shifted as compared to its absorption maximum in water, and it is similar to that of the excitation maximum observed in organic solvents. The excitation spectrum was found to be independent of emission wavelength in all the media studied. Similarly, the emission spectrum is also independent of excitation wavelength except that the fluorescence intensity is reduced when excitation is done at a wavelength other than the absorption maximum.
 | ||
| Fig. 1 Fluorescence spectra of diene 6 in different organic solvents and water at 298 K: (a) heptane, (b) 1,4-dioxane, (c) THF, (d) MeCN, (e) MeOH, (f) DMF and (g) water. Spectra are normalised at the maximum fluorescence intensity. | ||
Φ
f decreases with increase in solvent polarity. The change in the excited state dipole moment of 6 was determined from Lippert–Mataga analysis using the equation:25,26νa−νf={[2(μe−μg)2/hca3]F(D,n)}, where νa−νf is the Stokes' shift, μe−μg=Δμ is the change in dipole moment, h is Planck's constant, c is the velocity of light, a is the Onsagar radius and F(D,n)=Δf=(D−1)/(2D+1)−(n2−1)/(2n2+1), where D is the dielectric constant and n is the refractive index of the solvent. The Onsager radius has been taken as approximately 9 Å, a value that has been used for a similar diene, 1-p-N,N-dimethylaminophenyl-4-p-nitrophenylbuta-(1E, 3E)-diene.27 The Lippert–Mataga correlation plot is characterised by a straight line with a slope of 10634 and an R2 value of 0.9528, where R is the linear correlation. The change in excited state dipole moment (Δμ) is determined to be 22.54 D, indicating the polar nature of the fluorescent excited state of 6.
The dependence of the Stokes’ shifts on solvent polarity has also been correlated with the empirical solvent polarity parameters ET(30) and π*. ET(30) is a solvent polarity parameter proposed by Dimroth, Reichardt and coworkers, based on the transition energy for the longest wavelength solvatochromic absorption band of the pyridinium-N-phenoxide betain dye.28,29 The ET(30) value for a solvent is defined as the transition energy of the dissolved betaine dye, measured in kcal mol−1, and is recognised as one of the most comprehensive solvent polarity scales. The π* scale, proposed by Kamlet, Abboud and Taft, is derived from solvent effects on the π–π* transition of a variety of nitroaromatics.29,30 These solvent polarity correlations were made in various organic solvents, including 1,4-dioxane--water mixtures. When ET(30) is plotted against the Stokes' shifts in nonpolar, polar aprotic and polar protic solvents, good correlations are not obtained. However, good correlations are obtained when ET(30) is plotted against the Stokes' shifts in 1,4-dioxane–water mixtures. This can be due to the specific hydrogen bond interactions between protic solvents and excited diene. The ET(30) scale is known to show a strong dependence on hydrogen bond interactions. This can be the reason for the relatively poor correlations observed when ET(30) is plotted against the Stokes' shifts in apolar and protic solvents together. The ET(30) correlation plot for eight data points is characterised by a straight line slope of 162.7 and an R2 value of 0.9874. On the other hand, reasonable correlations with π* in aprotic as well as in protic solvents is observed. The π* correlation plot for eleven data points is characterised by a straight line slope of −0.0004 and an R2 value of 0.8326. Apparently, the fluorescent excited state of 6 has polar character and it undergoes hydrogen bond interactions with protic polar solvents.
The blue-shifted fluorescence emission of 6 in non-polar solvents can be from the locally excited state, while the longer wavelength emission can be from an intramolecular charge transfer excited state, which is more polar and lower in energy than the locally excited state. The intramolecular charge transfer excited state can involve conformational relaxation of the single bonds connecting the donor group to the acceptor group. Hence, the polar solvents stabilise the excited state, resulting in red-shifted fluorescence emission. The conformational relaxation of the excited state is possible by bond rotation at a number of sites, including carbon-carbon bonds between the aromatic centres as well as the bond between the donor dialkylamino group and the aromatic ring.
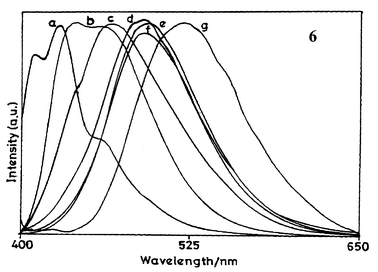
Low temperature fluorescence studies
The fluorescence spectral features of 6 in a 1∶1 (v/v) ethanol–methanol mixture at 298 and 77 K are shown in Fig. 2 and the corresponding data are summarised in Table 3. An examination of this data reveals that as compared to emission at 298 K, the emission of 6 is blue-shifted by 56 nm at 77 K. At 298 K, the diene exhibits fluorescence emission at 485 nm with a shoulder peak at 433 nm whereas at 77 K there is total disappearance of longer wavelength emission and the spectrum is characterised by well-defined structure with a maximum at 429 nm. Furthermore, as compared to 298 K, there is an approximately eight-fold increase in Φf at 77 K. The low temperature fluorescence observations point towards conformational relaxation in the excited state of diene 6. In a rigid matrix at 77 K, molecular motions and related geometrical changes in the fluorophores are expected to be reduced. This will result in a minimisation of solvent effects on the excited state. In the absence of solvent effects, a blue shift in the fluorescence emission is expected. At low temperature, non-radiative transitions are expected to be weak, hence the increase in Φf. It may, however, be mentioned that several other factors like a change in solvent density, a decrease in the population of some vibrational states, etc. may also need to be considered for a complete description of the blue shift observed at low temperature.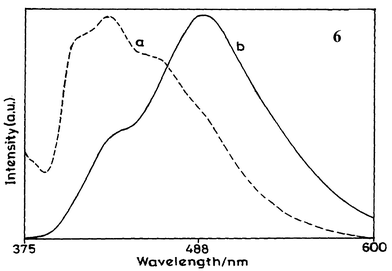 | ||
| Fig. 2 Fluorescence spectra of diene 6 in 1∶1 (v/v) methanol-ethanol matrix at (a) 77 K and at (b) 298 K. | ||
| T/K | λ ex max/nm | λ f max/nm | Φ f |
|---|---|---|---|
| 298 | 356 | 485, 433sh | 0.052 |
| 77 | 366 | 429 | 0.425 |
It is also interesting to note that in heptane and 1,4-dioxane at 298 K the intensity of the shorter wavelength fluorescence emission band is greater than that of the longer wavelength fluorescence emission band. In THF and ethanol–methanol mixtures, however, the intensity of the longer wavelength fluorescence emission band becomes much greater. Also, the fluorescence emission spectrum in methanol is characterised by the presence of only the CT band whereas that in 1∶1 ethanol–methanol is characterised by the presence of dual fluorescence emission bands.
It seems that the protic solvents influence the excited species more strongly when compared to the aprotic solvents. In protic solvents the hydrogen bond interactions may be important, affecting the fluorescence excited state and, hence, the fluorescence emission spectra. At low temperature in an ethanol–methanol (1∶1) matrix the excited state of diene is expected to have conformationally restricted geometries. On the other hand, at ambient temperature the excited diene can undergo conformational relaxation and occur in geometries that are different from the excited state geometries at low temperature. Therefore, the interactions between solvent and excited diene are expected to be different at the two temperatures. This may also contribute to the observed fluorescence excitation and emission features of 6.
Furthermore, in solvents having a dielectric constant (ε) greater than 30, such as methanol (ε=33), acetonitrile (36.6), DMF (38.3), and water (80) only the longer wavelength fluorescence emission band is seen for 6. However, two fluorescence bands for 6 are observed in a 1∶1 ethanol–methanol mixture. This may be due to the intermediate polarity of the mixed alcohols. The dielectric constant of ethanol is 24 and that of methanol is 33. So, a 1∶1 mixture of ethanol and methanol is expected to have dielectric constant between 24 and 33. Thus, while in solvents of high dielectric constant only the CT band is observed, in solvents of relatively lower dielectric constant (∼<30) both CT as well as non-CT bands appear.
Electronic absorption and fluorescence properties of the protein conjugates 8 and 9
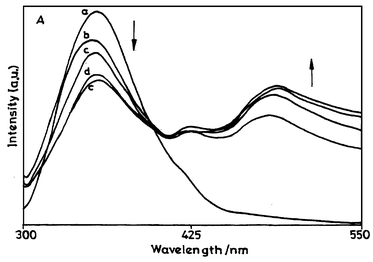
The UV-vis absorption and fluorescence spectra of BSA and the corresponding protein conjugate 8 with diene 6 are presented in Figs. 3 and 4, respectively. The fluorescence excitation spectra of diene 6 and its conjugate with BSA (8) are shown in Fig. 5. HSA showed similar absorption and fluorescence spectra. These absorption and fluorescence data are summarised in Table 4. | ||
| Fig. 3 UV-vis absorption spectra of (a) BSA and (b–e) protein conjugates in phosphate buffer. The concentration of diene in the conjugates is (b) 2.7×10−4, (c) 5.0×10−5, (d) 7.7×10−5 and (e) 8.8×10−5 M. | ||
 | ||
| Fig. 4 Fluorescence emission spectra of BSA and the corresponding protein conjugate in phosphate buffer: (a) pure BSA and (b–e) protein conjugates. The concentration of diene in the conjugates is (b) 2.7×10−5, (c) 5.0×10−5, (d) 7.7×10−5 and (e) 8.8×10−5 M. (λex
= 295 nm.) | ||
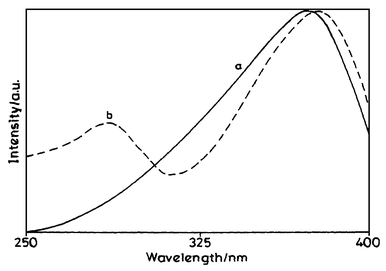 | ||
| Fig. 5 Excitation spectra of (a) diene 6 and (b) protein conjugate 8 in phosphate buffer (emission wavelength 478 nm). | ||
| Protein Conjugate | λ abs max/nm | λ ex/nm | λ f max/nm | λ ex max/nm | Φ f |
|---|---|---|---|---|---|
| a The values correspond to the maximum number of diene molecules bound to the protein, i.e., 1∶4.1 for 8 and 1∶3.88 for 9. | |||||
| 8 | 280 | 295 | 342, 478 | 280, 372 | 0.098 |
| 325, 365sh | 325 | 477 | 372, 280 | ||
| 365 | 478 | 372, 280 | |||
| 9 | 277 | 295 | 342, 476 | 279, 372 | 0.070 |
| 325, 368sh | 325 | 475 | 373, 280 | ||
| 365 | 476 | 373, 280 | |||
Conjugates 8 and 9 show absorption bands at around 280 nm (due to the protein part) and at 325 with a shoulder at around 365–368 nm due to the diene moiety. Thus, the absorption of the diene in the protein medium is blue-shifted as compared to its absorption in both organic solvents. We have noted that the absorption of diene 6 in aqueous medium is drastically blue-shifted. The relatively short wavelength absorption of 6 in water (310 nm as compared to an ∼370 nm absorption by 6 in organic solvents) can be due to the poor solubility of 6 in water and also due to ground state hydrogen bond interactions between 6 and water. Since the diene is not freely soluble in water, there is a chance of aggregation, which can reduce the absorption wavelength. Therefore, it can be said that in the protein conjugates 8 and 9, the coupled diene 6 is partially exposed to a relatively protic polar environment where it can participate in hydrogen bond interactions, resulting in a blue shift of its absorption maximum. This is also evident from the absorption maximum of 6 in methanol (354 nm) in which there is possibility of hydrogen bonding between the diene and the solvent.
The number of diene molecules coupled to BSA and HSA was determined by following the absorbance of the modified protein at 325 nm (ε 18400 mol−1 cm−1 l in 80% phosphate buffer+20% methanol) due to the diene moiety and 280 nm due to the protein residues. The absorption coefficients, ε, for BSA and HSA in phosphate buffer at 280 nm are 39080 and 36600 mol−1 cm−1 l, respectively. The relative concentrations of diene and protein moieties were calculated as discussed in the Experimental and it was found that a maximum of about four diene units could be coupled to BSA and HSA. It may be mentioned here that the spectra of the conjugates were taken in pure buffer, but the reference spectra with extinction coefficients are taken in a mixed solvent (20% methanol). As the compound is not properly soluble in water, its extinction coefficient in pure water could not be calculated. The extinction coefficient, therefore, was obtained using water containing 20% methanol, in which 6 is properly soluble. As said earlier there is a strong dependence of the absorption on alcohol mixture and therefore the values may not be the real ones that one expects in pure water.
Both the protein conjugates show similar fluorescence spectral characteristics, indicating that the diene is located in the same region in both the proteins. There is no change in the fluorescence emission or excitation maximum with increase in the concentration of the diene in the conjugates. However, there are changes in the fluorescence intensity. The fluorescence spectra of conjugates 8 and 9 were recorded by exciting the conjugates at 295, 325 and 365 nm. The 295 nm excitation (instead of 280 nm) was chosen since the excitation at 280 nm (an absorption band shown by the conjugates due to protein) can result in fluorescence emission by tyrosine residues of the protein as well. Excitation of conjugates 8 and 9 at 295 nm resulted in fluorescence emissions at 342 and 476–478 nm. Similarly, excitations of the conjugates at 325 and 375 nm resulted in fluorescence emission with maximum in the range of 475–478 nm. Thus, BSA and HSA both show fluorescence emissions at 342 nm when excited at 295 nm. Therefore, it can be said that the fluorescence emission of protein conjugates 8 and 9 at 342 nm is from the tryptophan residue of the modified proteins and the fluorescence emission at 478 nm is due to the diene moiety. However, at 295 nm the diene has considerable absorption and hence it can fluoresce. Therefore, the fluorescence emission of the diene was examined by exciting at 275 nm where it has a minimum in the absorption. Under these excitation conditions, two emission bands located at 342 and 478 nm were observed. This points towards the possibility of energy transfer interactions between tryptophan and diene moieties in the protein conjugates.
With an increase in the concentration of the diene moieties in the protein conjugate, there is a partial quenching of the intrinsic fluorescence of the protein at 342 nm (due to tryptophan residues) and an increase in the fluorescence emission at 478 nm (due to diene moieties) when excited at 295 nm. Since the diene also shows some absorption at 295 nm, an increase in the fluorescence intensity at 478 nm with increase in the concentration of diene molecules can be attributed to the excitation of the diene. However, there is a decrease in the fluorescence intensity of tryptophan with increase in the concentration of diene. This indicates that the diene causes quenching of the fluorescence due to tryptophan residues. To determine whether energy transfer was taking place from the tryptophan residues to the diene units, the fluorescence excitation spectrum of the protein conjugates 8 and 9 (obtained at 478 nm fluorescence emission) was compared with that of the fluorescence excitation spectrum of the diene in phosphate buffer (Fig. 5). The excitation spectrum of diene 6 is characterised by a band at 370 nm. However, the excitation spectrum of the protein conjugates featured bands at 280 as well as 370 nm, clearly indicating the involvement of the protein residue. These observations indicate the possibility of energy transfer from the tryptophan to diene moieties in both the conjugates.
Compared to the spectra in aqueous medium, the diene in the protein environments shows blue-shifted fluorescence emission and an increase in Φf. These observations indicate that the diene is located in a relatively non-polar environment in the protein. The fluorescence emission of diene in the protein conjugates is similar to that observed for the diene in medium polar solvents like THF in which the diene fluoresces at around 475 nm with a shoulder at 445 nm.
BSA and HSA are homologous proteins composed of single polypeptide chains with 583 and 585 amino acids, respectively, with a similar sequence and a similar conformation.31 The three-dimensional structure of HSA reveals that it contains three homologous domains that assemble to form a heart-shaped molecule.32–34 Each domain is the product of two sub-domains (A and B) that possess common structural motifs. It is also believed that the principle regions of ligand binding to HSA are located in hydrophobic cavities of sub-domains IIA and IIIA. However, the well-known difference between BSA and HSA is that while BSA has two tryptophans, HSA has only one tryptophan unit.
The tryptophan residue plays an important role as a chromophore and a fluorophore in optical studies of proteins. However, it is generally difficult to examine the behaviour of individual tryptophans in relation to the total conformational change of the protein, which contains the tryptophan(s), since quite a few tryptophans are located at random in most of the proteins. In this way, since there is only one tryptophan (Trp-214) unit present in HSA, which has been proposed to be located in a cleft or hydrophobic fold of sub-domain IIA, it provides a unique opportunity to study the energy transfer interactions and binding sites of the protein. In addition, one of the two tryptophans in BSA (Trp-213) is anticipated to be located in a similar environment as the single tryptophan residue (Trp-214) in HSA, while the additional tryptophan residue (Trp-134) has been proposed to be located on the surface of the molecule. The diene moiety can be located in the hydrophobic environment of sub-domain IIA of HSA, and in view of the observed similarity between the fluorescence characteristics of protein conjugates 8 and 9, it can be proposed that the diene moiety in BSA is also located in sub-domain IIA of the protein.
Conclusions
In conclusion, diene 6 exhibits solvatochromic fluorescence emission due to a polar, conformationally relaxed, intramolecular charge transfer excited state. The fluorescence behaviour of 6 can be used to investigate the micropolarity, binding site and energy transfer interactions in proteins. Thus, the present studies provide new directions for the development of fluorescence probes, based on charge transfer diphenylpolyenes, for organised structures in chemistry and biology.Acknowledgements
Research grant 37/7/95-R&D-II/559 from the Board of Research in Nuclear Sciences, Department of Atomic Energy, Government of India is gratefully acknowledged. We thank Dr. V. J Rao for providing diphosgene. We are also gratefully thankful to the reviewers of this paper for their valuable suggestions.References
- H. R. Horton and D. E. Jr. Koshland, Methods Enzymol., 1967, 11, 856 CAS.
- G. M. Edelman and W. O. McClure, Acc. Chem. Res., 1968, 1, 279.
- J. Reynolds, S. Herbert and J. Steinhardt, Biochemistry, 1968, 7, 1357 CrossRef CAS.
- A. Jonas and G. Weber, Biochemistry, 1971, 10, 1335 CrossRef CAS.
- B. Honore and P. C. Frandsen, Biochem. J., 1986, 236, 365 CAS.
- D. J. Cowley, Nature (London), 1986, 319, 14 CrossRef.
- S. S. Lehrer, Biochemistry, 1971, 10, 3254 CrossRef CAS.
- T. A. Wells, M. Nakazawa, K. Manabe and P. S. Song, Biochemistry, 1974, 33, 708.
- H. Bruderlein and J. Bernstein, J. Biol. Chem., 1979, 254, 11570 CAS.
- D. C. Turner and L. Brand, Biochemistry, 1968, 7, 3381 CrossRef CAS.
- E. Daniel and G. Weber, Biochemistry, 1966, 5, 1893 CrossRef CAS.
- A. Sktnik, D. Gormin and M. Kasha, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 11968 CAS.
- F. Moreno, M. Cortijo and J. Gonzalez-Jimenez, Photochem. Photobiol., 1999, 69, 8 CAS.
- T. Tsuchida, G. Zheng, R. K. Pandey, W. R. Potter, D. A. Bellnier, B. W. Henderson, H. Kato and T. J. Dougherty, Photochem. Photobiol., 1997, 66, 224 CAS.
- W. Rettig and R. Lapouyade, Fluorescence Probes Based on TICT States and Other Adiabatic Photoreactions, inTopics in Fluorescence Spectroscopy, ed. J. R. Lakowicz, vol. 4, Probe Design and Chemical Sensing, Plenum Press, New York, 1994 Search PubMed.
- A. K. Singh, D. Manjula and S. Kanvah, New J. Chem., 1999, 23, 1075 RSC.
- A. K. Singh and S. Kanvah, New J. Chem., 2000, 24, 639 RSC.
- A. K. Singh, D. Manjula and S. Kanvah, J. Phys. Chem. A, 2000, 104, 464 CrossRef CAS.
- A. K. Singh and G. R. Mahalaxmi, Photochem. Photobiol., 2000, 71, 387 CAS.
- A. K. Singh and D. Manjula, Biochim. Biophys. Acta, 2002, 1563, 35 CrossRef CAS.
- W. H. Melhuish, J. Phys. Chem., 1961, 65, 229 CrossRef CAS.
- S. R. Meech and D. Phillips, J. Photochem., 1983, 23, 193 CrossRef CAS.
- C. H. Self and S. Thompson, Nature (Medicine), 1996, 2, 817 Search PubMed.
- M. A. Brinkley, inPerspectives in Bioconjugate Chemistry, ed. C. F. Meares, American Chemical Society, Washington, D. C., 1993, pp. 59–70 Search PubMed.
- E. Lippert, Z. Electrochem., 1957, 61, 962 Search PubMed.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1956, 29, 465 CAS.
- D. C. Terauchi and T. Kobayashi, Chem. Phys. Lett., 1987, 137, 319 CrossRef.
- K. Dimroth, C. Reichardt, T. Siepmann and F. Bohlmann, Justus Leibigs Ann. Chem., 1963, 661, 1 CrossRef CAS.
- C. Reichardt, Angew. Chem., Int. Ed. Engl., 1965, 4, 29 CrossRef.
- M. J. Kamlet, J.-L. Abboud and R. W. Taft, J. Am. Chem. Soc., 1977, 99, 6027 CrossRef CAS.
- D. C. Carter and J. X. Ho, Adv. Protein Chem., 1994, 45, 153 CAS.
- D. C. Carter, X. M. He, S. H. Munson, P. D. Twigg, K. M. Gernert, M. B. Broom and T. Y. Miller, Science, 1989, 244, 1195 CAS.
- X. M. He and D. C. Carter, Nature (London), 1992, 358, 209 CrossRef.
- S. Sugio, A. Kashiara, S. Mochizuki, M. Noda and S. Kobayashi, Protein Eng., 1999, 12, 439 CrossRef.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2004 |