Preparation, characterisation and complexation of a redox-active, soft-donor macrocycle
Hilary J.
Hartigan
,
Georg
Seeber
,
Andrew R.
Mount
,
Lesley J.
Yellowlees
and
Neil
Robertson
*
School of Chemistry, University of Edinburgh, West Mains Road, Edinburgh, UK EH9 3JJ. E-mail: neil.robertson@ed.ac.uk; Fax: 0131 6504743; Tel: 0131 650 4755
First published on 31st October 2003
Abstract
Two new redox-active macrocycles 2 and 3 have been prepared based on an 18-membered S4N2–donor ring conjugated to one or two CpCo(dithiolene) redox/chromophore units respectively. Cyclic voltammetry for 2 showed one reversible reduction process at −0.29 V and EPR spectroscopy of the reduced form indicated a significant degree of delocalisation of the unpaired electron. The compound 2 showed an intense low-energy absorption at 675 nm (in CH2Cl2) that was shown to shift to higher energy during electrochemical reduction. Interaction of 2 with guest Ag(I) ions was followed by monitoring changes in the electrochemical and spectroscopic behaviour with sequential additions of the guest metal ion and by electrochemistry at a silver electrode. UV/Vis spectroscopic results indicated a strong interaction of Ag(I) with 2 involving large changes in the electronic nature of the macrocycle chromophore. Cyclic voltammetry also indicated interaction between 2 and Ag(I) and suggested the involvement of a film containing 2 on the electrode surface.
Introduction
Macrocyclic ligands possessing properties such as visible absorption, emission and reversible redox behaviour have been widely studied as sensors for metal ions. Much research has been carried out where alteration of these physical properties can be observed in the presence of metal ions. This can be used in metal-ion detection and practical electrochemical and optical sensors have emerged from this field.1 Importantly, the ability of macrocycles to show selectivity in their binding ability towards metal ions2,3 offers tremendous potential in the fabrication of specific sensors for selected ions. To date however, work in this field has overwhelmingly been based on complexation studies involving the binding of alkaline earth and alkali metal ions to hard donor systems such as crown ethers and mixed O and N-donor systems. These have been the subject of several reviews.1,4,5In contrast, only a very small amount of research has been carried out on the complexation behaviour of transition metal or heavy metal ions with soft-donor coloured, fluorescent6 or redox-active macrocycles. Such systems however, would be expected to show enhanced binding capabilities and selectivity for softer heavy metals and transition metals in comparison with oxygen and nitrogen-donor systems. The limited number of examples reported in this area are based around a few main redox systems including redox-active units such as tetrathiafulvalene (TTF) linked with thioether crowns,7–10 ferrocene units with thioether11 and mixed S and N or N and O donor macrocycles,12,13 porphyrazines,14 and other transition metal based systems.15 Despite the inclusion of redox centres in these receptors, only the studies involving TTF-based systems made much use of electrochemical methods to investigate the binding of guest ions in the macrocycle. Examples have also been reported involving metal bis-1,2-dithiolene complexes as the redox-active component of the molecule.16,17 In these however, the strength of interaction between the guest metal ion and the redox center is typically small, due to intervening aromatic groups which reduce communication and/or a poor interaction between soft sulfur donors and hard metal cations.
Soft-donor, redox-active macrocycle systems have significant relevance in both industrial and environmental areas, with the potential of such compounds to sense environmentally toxic metals. Despite this however, the above examples remain relatively isolated and little systematic study of soft-donor, redox-active macrocycles has been carried out to assess the selectivity of these systems to a range of transition and heavy metal cations.
In order to develop new redox-active systems with soft donor atoms, we report the preparation and study of new cobalt based redox-active macrocycles based on the macrocyclic ligand 1
(Scheme 1).18 These involve the CpCodithiolene (Cp![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =cyclopentadienyl) unit that has been previously shown to possess the reversible redox activity and characteristic chromophore19 sought in the new macrocycles presented here. By incorporating dithiolene ligands into the system we aim to achieve the high degree of delocalisation vital for the significant interaction of the host and guest metal centres. Thus, sulfur atoms that form part of the electronically-delocalised redox-active/chromophore unit are also potential donor atoms within the macrocyclic ring to maximise the guest ion perturbation on the electronic properties of the host. Synthesis and characterisation of the new macrocycles is described and electrochemical and spectroscopic complexation studies of one of these redox-active macrocycles with Ag(I) is presented and discussed.
=cyclopentadienyl) unit that has been previously shown to possess the reversible redox activity and characteristic chromophore19 sought in the new macrocycles presented here. By incorporating dithiolene ligands into the system we aim to achieve the high degree of delocalisation vital for the significant interaction of the host and guest metal centres. Thus, sulfur atoms that form part of the electronically-delocalised redox-active/chromophore unit are also potential donor atoms within the macrocyclic ring to maximise the guest ion perturbation on the electronic properties of the host. Synthesis and characterisation of the new macrocycles is described and electrochemical and spectroscopic complexation studies of one of these redox-active macrocycles with Ag(I) is presented and discussed.
 | ||
| Scheme 1 Preparation of new CpCodithiolene-containing macrocycles. L=cyclooctadiene (COD) or (CO)2. | ||
Results and discussion
(i) Preparation of the new macrocycles
Two new highly-coloured, redox-active, soft-donor macrocycles were prepared by reaction of the dithione S4N2 donor macrocycle 1 with CpCoL (L=COD, (CO)2)
(Scheme 1). The most successful preparation involved reaction of 1 with two equivalents of CpCo(CO)2, for three hours in THF. Purification involved simple chromatographic procedures and led to the high-yield isolation (62%) of the mono-substituted CpCodithiolene–S4N2 donor macrocycle 2 as a dark green microcrystalline powder.
The procedure was based on a literature method for related CpCodithiolene complexes,20 which reports the use of CpCoCOD as the ‘CpCo’-source, the higher boiling point solvent toluene and reaction conditions of 48 hours reflux. We employed THF as the solvent as we found that a lower reflux temperature gave a higher yield, and CpCo(CO)2 was used as an alternative reagent to CpCoCOD as it showed more rapid formation of the product at the lower temperature used.
The literature conditions for related reactions20 led to a mixture of the mono-and di-substituted CpCodithiolene–S4N2 macrocycles 2 and 3 in 8% and 10% yields respectively with poorer purity. Yields of complex 3 were low under all of the conditions studied and only fundamental characterisation was carried out for this complex. Complex 2 was prepared by the optimised conditions above and purified by chromatography. The increased quantity available allowed detailed characterisation including spectroelectrochemistry, cyclic voltammetry, EPR and guest-ion binding studies and these are detailed below.
Numerous attempts were made to grow crystals of 2 by a variety of crystallisation techniques, however all were unsuccessful and the observation of degradation products suggest that complex 2 decomposes over prolonged periods in solution. A published structure for the precursor 1 has previously shown the geometry of the macrocycle unit.18
(ii) Spectroscopic and electrochemical characterisation of 2
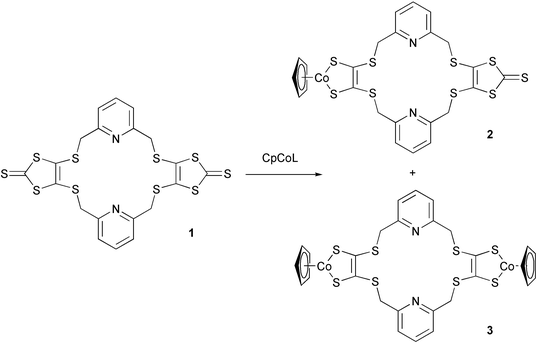
Cyclic voltammetry for 2 was carried out (Fig. 1) and showed a reversible one-electron reduction at E![[circle, cut, short horiz bar]](https://www.rsc.org/images/entities/char_e0eb.gif) =−0.29 V with an anodic to cathodic peak separation of 63 mV. The extended delocalisation within the system limits the usefulness of metal oxidation state to describe the complex, however for convenience the process will be designated as a Co(III)/Co(II) reduction where the Cp is formally regarded as a monoanion and the dithiolene as a dianion.21 Electrogeneration of the reduced species, by holding the potential at −0.40 V, resulted in a colour change from dark green to reddish/brown and the number of coulombs passed confirmed this process as a one-electron transfer. The dark green Co(III) species could be regenerated by electrochemical oxidation at 0 V. The Co(III)/Co(II) couple was shown to be electrochemically reversible at all scan rates. A chemically irreversible oxidation process with a peak potential of +1.2 V was also observed and assigned to oxidation of the pyridyl groups on the macrocyclic ring.
=−0.29 V with an anodic to cathodic peak separation of 63 mV. The extended delocalisation within the system limits the usefulness of metal oxidation state to describe the complex, however for convenience the process will be designated as a Co(III)/Co(II) reduction where the Cp is formally regarded as a monoanion and the dithiolene as a dianion.21 Electrogeneration of the reduced species, by holding the potential at −0.40 V, resulted in a colour change from dark green to reddish/brown and the number of coulombs passed confirmed this process as a one-electron transfer. The dark green Co(III) species could be regenerated by electrochemical oxidation at 0 V. The Co(III)/Co(II) couple was shown to be electrochemically reversible at all scan rates. A chemically irreversible oxidation process with a peak potential of +1.2 V was also observed and assigned to oxidation of the pyridyl groups on the macrocyclic ring.
 | ||
| Fig. 1 Cyclic voltammogram of 2 in dichloromethane containing 0.4 mol dm−3 TBABF4 at 25°C under N2. | ||
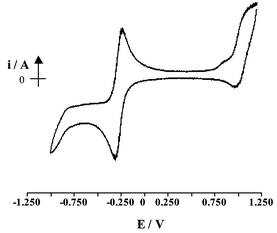
Electrogeneration of compound [2]− was carried out, whilst monitoring the process using in situ UV/Vis spectroscopy. A decrease in the bands due to 2 {A, C, E and G} was observed along with sequential growth of those due to [2]− (B, D and F) (Fig. 2). The molar extinction coefficients (ε) and assignments for each band are reported in Table 1.
![Electrochemical reduction of 2 in CH2Cl2–0.4 M [TBA][BF4] followed by electronic spectroscopy.](/image/article/2004/NJ/b309317d/b309317d-f2.gif) | ||
| Fig. 2 Electrochemical reduction of 2 in CH2Cl2–0.4 M [TBA][BF4] followed by electronic spectroscopy. | ||
| 2 | [2]− | ||||
|---|---|---|---|---|---|
| λ max/nm (cm−1) | ε/ M−1cm−1 | λ max/ nm (cm−1) | ε/ M−1cm−1 | ||
| A | 288 (34749) |
32756 |
B | 290 (34440) |
21009 |
| C | 389 (25687) |
11747 |
D | 420 (23834) |
12651 |
| E | 430 (23267) |
13328 |
|||
| G | 675 (14978) |
17620 |
F | 506 (19766) |
11973 |
The band at 675 nm for 2 was assigned as arising from a HOMO–LUMO transition involving delocalised orbitals comprising both metal and ligand orbital character. Little solvatochromism was observed for this band (for example the transition was observed at 664 nm in DMF), supporting the view that both metal- and ligand-based orbitals are involved in both the HOMO and LUMO and that little charge-transfer takes place. The band which grows in at 506 nm upon reduction from Co(III) to Co(II), was assigned as the corresponding Co(II) band. The higher energy for this transition is consistent with the partial occupation of the LUMO upon reduction making transition into this orbital higher in energy. All other absorbances in the spectrum of 2 are present in the UV/Vis spectrum of the precursor macrocycle 1. Thus all higher bands can be assigned to π–π* transitions within the macrocyclic ligand.
The EPR spectrum for the CoII species [2]−, generated by electrochemical reduction, was obtained in fluid solution at room temperature and in a frozen glass at 77 K. The room temperature spectrum showed the expected 59Co (I=7/2) octet with gav=2.105 and hyperfine coupling constant Aiso=38.5 G. The frozen-solution spectrum of [2]−
(Fig 3) indicated a rhombic system where all three axes are inequivalent, consistent with the geometry of the molecule. A computer simulated spectrum allowed the determination of the relevant parameters giving g values of 2.246, 2.043 and 1.993 with corresponding A values of 89, 15.5 and 30 G and line widths of 12, 10 and 8 G.
![Experimental EPR spectrum of reduced macrocycle [2]− in CH2Cl2–0.5 M TBABF4 at 77K; frequency 9.47 GHz.](/image/article/2004/NJ/b309317d/b309317d-f3.gif) | ||
| Fig. 3 Experimental EPR spectrum of reduced macrocycle [2]− in CH2Cl2–0.5 M TBABF4 at 77K; frequency 9.47 GHz. | ||
The coupling constants observed are smaller than typical for Co(II) radicals, and this is indicative of a system in which the unpaired electron is delocalised over both the metal and the ligand environment.22–25 The EPR data for related CoII-dithiolene-based compounds previously reported in the literature22,23 have also shown similarly small coupling constants. These include {Co(NO)[S2C2(CF3)2]2}− where Aiso is reported as 32.9 G and gav as 2.059 and {Co(NO)(S2C2Ph2)2]− with an Aiso of 29.4 G and a gav of 2.050.24 In both cases, the highly delocalised ligand system directly attached to the Co results in the lower coupling values for these compounds. In the case of [2]−, a similar effect was observed and this reflects the influence of both the cyclopentadienyl moiety and the dithiolene group, both of which are highly delocalised. This suggests delocalisation of the LUMO of 2 over much of the complex and is consistent with the results observed in the UV/Vis spectrum.
(iii) Complexation studies
Work was aimed towards assessing the use of 2 for the sensing of guest metal ions by monitoring both the spectroscopic and the electrochemical response to the stepwise addition of metal ions. This report includes an investigation of the binding of Ag(I) as this was expected to interact strongly with the soft-donor system of 2. The sequential addition of aliquots of a solution of Ag(I) to a solution of 2 was followed using both UV/Vis spectroscopy and cyclic voltammetry and these are discussed below.The UV/Vis spectral changes upon the addition of one equivalent of Ag(I)ClO4 to a solution of 2 in DMF are shown in Fig. 4.
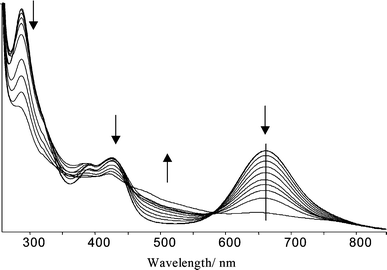 | ||
| Fig. 4 Sequential addition of 0.1 equivalents of Ag(I)ClO4 to 2 in DMF followed by UV/Vis spectroscopy. The vertical line is a guide to the eye. | ||
An extremely large decrease in intensity of the absorbance at 664 nm was observed along with a shift in the peak wavelength of 20 nm (468 cm−1) from 664 to 644 nm and the growing in of a broad new absorbance at around 500 nm. These indicate a large perturbation in the electron density of the CpCodithiolene chromophore. Such a change is taken to be a result of the complexation of the guest silver ion to the macrocycle. Thus the UV/Vis data suggest binding between the Ag(I) guest ions and the host cavity of the macrocycle 2 in such a way that direct interaction takes place between the guest ion and the donor sulfur atoms that also comprise part of the CpCodithiolene chromophore unit.
The collapse of the CpCodithiolene absorbance at 664 nm was in this case observed to proceed almost to completion with 1 equivalent of Ag(I)ClO4 added. Any further additions showed no further significant changes in the intensity of this absorbance. Complexation proceeds through two clear stages. The first involves addition of the Ag(I)ClO4 salt from 0–0.5 equivalents where the absorbance at 664 nm was found to decrease to approximately half the initial intensity (Fig. 5a). A clear set of isosbestic points can be seen at 277, 320 and 585 nm during this change. Consideration of the absorbances observed at 288, 389 and 430 nm indicates that up to 0.5 equivalent additions causes little change in their intensity. During additions of Ag(I)ClO4 from 0.6–0.9 equivalents (Fig. 5b), a different set of isosbestic points was observed that were not coincident with those recorded during addition of the first 0.5 equivalents of Ag(I). Each addition following 0.5 equivalents caused the absorbances at 288, 389 and 430 nm to decrease much more significantly in intensity. A possible explanation for the two differing sets of isosbestic points involves the initial formation of a species Ag(I)·[2]2, with further addition of Ag(I) giving Ag(I)·[2]. Addition of the final 0.1 equivalent of Ag(I) led to a spectrum that did not exactly coincide with the isosbestic points suggesting that the onset of some further new interaction between (Ag(I)[2]) had begun due to the presence of a slight excess of Ag(I).
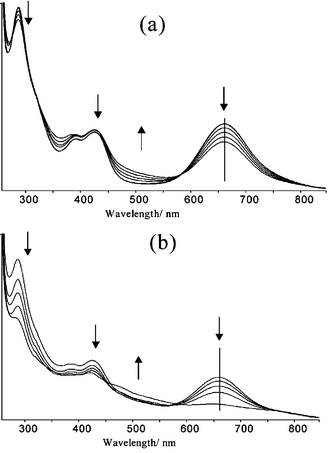 | ||
| Fig. 5 (a) Addition of 0–0.5 equivalents of AgClO4 to 2. (b) Addition of 0.6–1.0 equivalents of AgClO4 to 2. | ||
Attempts were made to derive a stability constant for formation of Ag(I)[2] from the change in the absorbance at 664 nm, however this was unsuccessful since the best-fit line through the points was essentially linear. This indicates that the value of the formation constant, K, between Ag(I) and 2 is too large to be determined by these data.
Cyclic voltammetry was carried out for the macrocycle 2 in THF using a silver wire as the working electrode. Scanning to negative potential showed the Co(II)/Co(III) redox couple at a potential (Fig. 6, cycle 1) comparable to that observed using the Pt electrode (Fig. 1). Extending the scanning range to +0.75 V resulted in the observation of a rising current attributable to stripping of Ag(I) from the electrode. The onset of this current was observed to occur 0.12 V less positive compared with the stripping current observed in the absence of 2, indicating stabilisation of the dissolved Ag(I) by complexation with 2.
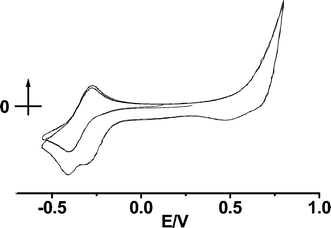 | ||
| Fig. 6 Cyclic voltammogram of 2 in THF at a silver working electrode. The first cycle, scanning to negative potential shows the Co(III)/Co(II) redox couple. In the second cycle, scanning is extended to give Ag(I) formation at positive potential resulting in new reduction processes. | ||
Several return reductions were observed to arise from this oxidation (Fig. 6, cycle 2), most notably a new reduction peak at −0.30 V. In addition, when compared with cycle 1, there is an appreciable current attributable to the reduction of Ag(I) throughout the region 0.75 to −0.30 V. Reduction over such a potential range could be indicative of a process limited by either slow, potential-independent reduction kinetics or reduction at a distribution of characteristic potentials. Slow kinetics can be discounted however, as after Ag(I) generation, either pausing the cycle at +0.5 V for 60 seconds, followed by resuming cycling or reducing the CV sweep rate did not significantly affect the CV response. It therefore seems that a distribution of Ag(I) reduction potentials are observed. Disconnecting the working electrode and resuming the sweep also had negligible effect demonstrating that the oxidised Ag(I) generated cannot be lost by diffusion and must remain bound to the electrode surface. This is good evidence for the formation of a surface film, presumably involving stabilisation of Ag(I) species through interaction with 2, involving a number of different coordination environments for Ag(I) leading to a range of reduction potentials. Some of the Ag(I) ions must be sufficiently stabilised by binding to 2 that they remain in the oxidised form until reduction at −0.30 V. The presence of a peak at −0.30 V suggests reduction of Ag(I) located in a specific and stable binding site (most likely the macrocyclic cavity) at this potential. The position of the redox peaks for 2 remain unchanged.
The strength of initial binding between 2 and Ag(I) can be estimated from the shift of −0.12 V of the oxidation of silver from the working electrode in the presence of 2. Since lnK=nF/RT(EAg(I)
−
E2.[Ag(I)]),26 the stability constant (K) for the initial complex formed between 2 and [Ag(I)] can be calculated as 102. This should be regarded as a lower limit however, as re-reduction of Ag(I) takes place over a wide and much more negative potential range, suggesting that the initial complex can rearrange to more stable species. Stability constants for complexes of Ag(I) with sulfur-donor or mixed sulfur-nitrogen-donor macrocycles have been reported as around K=1012 to 1014.27 These results would therefore suggest initial binding of 2 to Ag(I) is followed by rearrangement to a more strongly bound complex.
These studies provide clear evidence for an interaction between Ag(I) and the macrocycle 2 and demonstrates that film formation involving 2 and Ag(I) on the silver wire electrode occurs and plays a significant role in the electrochemical behaviour of the system.
Electrochemical experiments were also carried out using a Pt disc electrode with sequential addition of a solution of Ag(I) to macrocycle 2 in THF. Following each addition, equilibration was allowed for 30 minutes and the cyclic voltammogram recorded (Fig. 7). A new reduction peak was observed to grow in at −1.29 V, which increased steadily with increasing Ag(I) concentration and is therefore assigned as reduction of Ag(I). Again, little change was observed of the initial Co(III)/Co(II) redox peaks of 2. Significantly, there was a lack of any normal stripping or reduction signal for silver metal, even in scans where a large excess of Ag(I) had been added to the solution. Instead, it was observed that when more than one equivalent of Ag(I) was added, the reduction at −1.29 V continued to increase and an increase was observed in the peak associated with the irreversible macrocycle oxidation at +1.2 V.
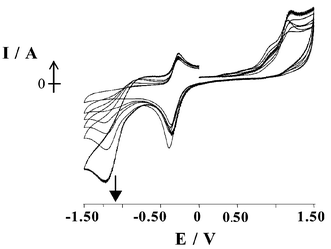 | ||
| Fig. 7 Sequential addition of Ag(I) to 2 in THF. | ||
These results suggest that the macrocycle 2 adsorbs onto the surface of the electrode, blocking access for added excess Ag(I) ions to the electrode. Such a surface film would have little effect on the appearance of the Co(III)/Co(II) redox peaks as mediated electron transfer via the surface Co(III)/Co(II) centres to the solution Co(III)/Co(II) is occurring, consistent with the observation that Ag(I) binding does not perturb the Co(III)/Co(II) redox potential. Reduction of Ag(I) however would be greatly affected by such a film and the lack of reduction processes for Ag(I) at potentials corresponding to those seen at the silver wire (Fig. 6) is presumably due to the alteration of the nature of the electrode surface and thus an increase in the energy for silver plating.
In order to further test this mechanism, an experiment was carried out where a film of 2 on a Pt electrode was deliberately prepared through drop coating. CV experiments were carried out, using CH3CN solvent since 2 is insoluble and the film will not be removed by dissolution. This experiment confirmed that redox peaks for uncomplexed Ag(I) can be effectively blocked by a film of 2 (Fig. 8). Although on the first cycle, both the surface film of 2 and solution Ag(I) displayed redox processes, by the third cycle only Co(II)/Co(III) redox peaks were observed at −0.3 and −0.1 V with Ag(I) plating and stripping peaks completely blocked. The requirement for three cycles to achieve blocking of the Ag peaks is presumably due to some redox-induced surface rearrangement of 2 to complete a coherent, pinhole-free surface film.
![Cyclic voltammogram of a film of 2 on a Pt electrode in a CH3CN solution containing Ag(i) ions. On the first cycle silver plating and stripping peaks are observed at 0.44 and 0.7 V respectively (highlighted in the box) in addition to Co(iii)/Co(ii) redox peaks at −0.3 and −0.1 V. By the third cycle only the Co(iii)/Co(ii) peaks were observed and the silver electrochemical processes were blocked by the film. (Note that the Co(iii)/Co(ii) redox peaks gradually decrease by redox film thinning through cycling probably due to some solubility of [2]−).](/image/article/2004/NJ/b309317d/b309317d-f8.gif) | ||
| Fig. 8 Cyclic voltammogram of a film of 2 on a Pt electrode in a CH3CN solution containing Ag(I) ions. On the first cycle silver plating and stripping peaks are observed at 0.44 and 0.7 V respectively (highlighted in the box) in addition to Co(III)/Co(II) redox peaks at −0.3 and −0.1 V. By the third cycle only the Co(III)/Co(II) peaks were observed and the silver electrochemical processes were blocked by the film. (Note that the Co(III)/Co(II) redox peaks gradually decrease by redox film thinning through cycling probably due to some solubility of [2]−). | ||
Conclusions
We have synthesised the first examples of metal complexes with a bis-1,2-dithiolene ligand extended to include a mixed S/N soft-donor macrocycle. These were designed to maximise interaction between the redox-active, highly-coloured CpCodithiolene unit and any metal ion bound as a guest in the macrocycle. Spectroscopic (UV/Vis, EPR and spectroelectrochemistry) characterisation of the redox-active macrocycle 2 were consistent with extended electronic delocalisation leading to the possibility of strong interaction between the redox centre and the guest ion. The potential of complex 2 as a redox or spectroscopic ion sensor was assessed by following changes in UV/Vis spectroscopy and cyclic voltammetry through sequential addition of Ag(I) cations. The UV/Vis study confirmed the anticipated strong perturbation on the electronic properties of the host macrocycle upon complexation of the guest ion. The electrochemical study, using both a silver and a platinum working electrode, clearly demonstrated that an interaction occurs between 2 and Ag(I) ions either generated from oxidation at the silver working electrode or added as a silver salt. At both platinum and silver working electrodes, formation of a film at the electrode involving 2 is implicit in the interpretation of the electrochemical results.Experimental
The macrocycle 118 was prepared according to the literature procedures. All solvents were purchased from Aldrich and were distilled and dried before use. Electrochemical studies were performed using a DELL 466DL personal computer with General Purpose Electrochemical System (GPES) Version 4.5 software connected to an Autolab system containing a PSTAT20 potentiostat. All of the electrochemical techniques used a three-electrode configuration. The reference electrode used was Ag/AgCl in a solution of 0.45 M [TBA][PF6] in MeCN against which E for the ferrocinium/ferrocene couple was measured to be +0.55 V. The working and counter electrodes were a platinum microdisc (0.5 mm diameter) and a large surface area platinum wire respectively. Coulometric experiments were performed in a conventional H-type cell using large surface-area Pt working and counter electrodes. All solutions were purged with dry nitrogen prior to electrochemical study. All cyclic voltammograms were recorded at a scan rate of 100 mV s−1 unless otherwise stated. The optically transparent thin-layer electrode (OTTLE) experiments were performed as described previously.28 All spectra were recorded on a lambda-9 spectrophotometer (Perkin Elmer) which was controlled by a Datalink PC running UV Win Lab software (version 2.70.01). EPR spectra were recorded on an X-band Bruker ER200D spectrometer using EPR acquisition system version 2.42 software. All EPR spectra were corrected for dpph, g=2.0036.
Preparation of the mono substituted CpCo–S4N2 donor macrocycle 2
The dithione S4N2 donor macrocycle 1 (3.47 g, 5.7 mmol), was dissolved in 50 ml of THF and the solution allowed to stir under an inert atmosphere of N2. To the reaction mixture was added a 10 ml solution of CpCo(CO)2 (2.07 g, 1.2 mmol). The reaction mixture was heated to reflux and the temperature maintained for three hours. Hot filtration of the reaction mixture gave a dark green filtrate, which was brought to dryness in vacuo. Column chromatography on silica eluting with 2 ∶ 1 hexane : CH2Cl2 gave a dark green solution. This was reduced to 40 ml in vacuo and stored in a freezer at −20°C for four days. Filtration delivered a light green solid of 2
(2.41 g, 62%).
Calc. for C24H19S9N2Co, C, 42.2; H, 2.8; N, 4.1%, found C, 42.8; H, 3.3; N, 3.7. IR (KBr)
ν(C–H) 3448(w), ν(py C–C) 1638(s), ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) 1451, ν(C–S) 1384(m), ν(CS) 1061(s). UV/Vis (CH2Cl2)
λmax=288 (ε=34749 M−1cm−1), 389 nm (25687 M−1cm−1), 430 nm (23267 M−1cm−1), 675 nm (14978 M−1cm−1). Electrospray MS [CH2Cl2]: 683 [M+]
C) 1451, ν(C–S) 1384(m), ν(CS) 1061(s). UV/Vis (CH2Cl2)
λmax=288 (ε=34749 M−1cm−1), 389 nm (25687 M−1cm−1), 430 nm (23267 M−1cm−1), 675 nm (14978 M−1cm−1). Electrospray MS [CH2Cl2]: 683 [M+]
Preparation of a mixture of the mono- and di- substituted CpCodithiolene–S4N2 macrocycles 2 and 3 respectively
The dithione S4N2 donor macrocycle 1 (0.26 g, 0.43 mmol) was dissolved in toluene (20 ml) and allowed to stir under an inert atmosphere of N2. To the reaction flask was added a 10 ml toluene solution of CpCoCOD (0.20 g, 0.86 mmol). The reaction mixture was heated to reflux and this temperature was maintained for 48 hours. The reaction mixture was cooled and 40 ml of dichloromethane added and heated to reflux again. Hot filtration delivered a dark green filtrate. The reaction mixture was brought to dryness in vacuo and the remaining solid was re-dissolved in CH2Cl2. Dry flash chromatography on silica, eluting with CH2Cl2 delivered a dark green solution. The volume was reduced down to 90–100 ml and stored in a freezer at −20°C for four days. Filtration delivered a light green solid of 3 in 10% yield. The dark green filtrate was reduced to 20–25 ml, hexane layered on and stored in a freezer at −20°C over night. Filtration delivered 2 as a dark green/black solid in 8% yield.
2 : Calc. for C24H19S9N2Co: C, 42.2; H, 2.8; N, 4.1% found, C, 41.1; H, 3.5; N, 3.8. FAB-MS (3-NOBA): 683 [M+].
3 : Calc. for C28H24S8N2Co2, C, 44.1; H, 3.2; N, 3.7%, found, C, 37.5; H, 2.8; N, 4.0. IR: (KBr)
ν(C–H) 3448(w), ν(py C–H) 1638(s), ν(Cp C–H) 1571(s), ν(CC) 1450(m). UV/Vis: 274 nm (ε=23137 M−1cm−1), 307 nm (17186 M−1cm−1), 382 nm (20342 M−1cm−1), 741 nm (4942 M−1cm−1). FAB-MS (3-NOBA): 763 [M+].
Complexation studies of Ag(I) to 2
Solutions (0.2 mM) of the macrocycle 2 (25 ml) were prepared in the appropriate solvent. In all cases the solutions contained 0.5 M [TBA][BF4], which acted as a background electrolyte for the electrochemical studies and minimised the influence of changes in ionic strength in the UV/Vis studies. The solvent employed was distilled and dried before use and was de-gassed prior to electrochemical study in order to eliminate any dissolved O2. Addition was made of equal portions (0.1 equivalents in 0.1 ml) of the Ag(I) salt dissolved in the given degassed solvent (also containing 0.5 M TBABF4). The electrochemical cell used was as described above. Following each addition of the metal ion salt, equilibration was allowed for 30 minutes. An analogous method was used to prepare solutions for UV/Vis spectra and these were recorded under N2 on extracted aliquots that were then reintroduced to the bulk solution.Acknowledgements
We thank the EPSRC for financial support and Dr Eric J. L. McInnes, University of Manchester for simulation of the frozen EPR spectrum.References
- Comprehensive Supramolecular Chemistry, vol. 1, ch. 16, 17, 18, 19, Elsevier Science Ltd., Oxford, 1996 Search PubMed.
- L. S. W. L. Sokol, L. A. Ochrymowyez and D. B. Rorabacher, Inorg. Chem., 1981, 20, 3189 CrossRef CAS.
- M. Kodama and E. Kimura, J. Chem. Soc., Dalton Trans., 1980, 2536 RSC; F. C. J. M van Veggel, W. Verboom and D. N. Reinhoudt, Chem. Rev., 1994, 94, 279 CrossRef.
- P. D. Beer, Chem. Soc. Rev., 1989, 18, 409 RSC.
- H-G. Löhr and F. Vögtle, Acc. Chem. Res., 1985, 18, 65 CrossRef.
- R. T. Bronson, J. S. Bradshaw, P. B. Savage, S. Fuangswasdi, S. C. Lee, K. E. Krakowiak and R. M. Izatt, J. Org. Chem., 2001, 66, 4752 CrossRef CAS.
- T. Jørgensen, B. Girmay, T. K. Hansen, J. Becher, A. E. Underhill, M. B. Hursthouse, M. E. Harman and J. D. Kilburn, J. Chem. Soc., Perkin Trans. 1, 1992, 2907 RSC.
- C. Gemmell, G. C. Janairo, J. D. Kilburn, H. Ueck and A. E. Underhill, J. Chem. Soc. Perkin Trans., 1994, 2715 Search PubMed.
- M. Wagner, D. Madsen, J. Markussen, S. Larsen, K. Schaumburg, K-H. Lubert, J. Becher and R-M. Olk, J. Chem. Soc. Perkin Trans. 1, 1996, 1995 RSC.
- N. Robertson, S. Vukojevic, X. Liu, L. J. Yellowlees and S. Parsons, J. Chem. Soc., Dalton. Trans., 1999, 3913 RSC.
- M. Sato, M. Katada, S. Nakashima, H. Sano and S. Akabori, J. Chem. Soc., Dalton Trans., 1990, 1979 RSC; M. Sato, S. Tanaka, S. Akabori and Y. Habata, Bull. Chem. Soc. Jpn., 1986, 59, 1515 CAS; M. Sato, S. Akabori, M. Katada, I. Motoyama and H. Sano, Chem. Lett., 1987, 1847 CAS; M. Sato, K. Suzuki and S. Akabori, Bull. Chem. Soc. Jpn., 1986, 59, 3611 CAS; M. Sato and S. Akabori, Bull. Chem. Soc. Jpn., 1989, 62, 3492 CAS.
- P. D. Beer, J. E. Nation, S. J. W. McWhinnie, M. E. Harman, M. B. Hursthouse, M. I. Ogden and A. H. White, J. Chem. Soc., Dalton Trans., 1991, 2485 RSC.
- J. M. Lloris, R. Martinez-Manez, T. Pardo, J. Soto and M. E. Padilla-Tosta, Chem. Commun., 1998, 837 RSC.
- S. L. J. Michel, B. M. Hoffman, S. M. Baum and A. G. M. Barrett, Prog. Inorg. Chem., 2001, 50, 473 CrossRef CAS.
- V. W-W. Yam, Y-L. Pui, W-P. Li, K. K-W. Lo and K-K Cheung, J. Chem. Soc., Dalton Trans., 1998, 3615 RSC.
- M. L. H. Green, W. B. Heuer and G. C. Saunders, J. Chem. Soc., Dalton Trans., 1990, 3789 RSC.
- N. D. Lowe and C. D. Garner, J. Chem. Soc., Dalton Trans., 1993, 3333 RSC.
- B. Girmay, J. D. Kilburn, A. E. Underhill, K. S. Varma, M. B. Hursthouse, M. E. Harman, J. Becher and G. Bojesen, J. Chem.Soc., Chem Commun, 1989, 1406 RSC.
- M. Fourmigue, Coord. Chem. Rev., 1998, 178–180, 823 CrossRef CAS.
- A. R. Siedle, J. Organomet. Chem., 1976, 120, 369 Search PubMed.
- M. Kajitani, T. Akiyama, A. Sugimori, K. Hirakata, Y. Hoshina, Y. Satsu, G. P. Sato and K. Shimizu, J. Electroanal. Chem., 1988, 251, 421 CrossRef CAS.
- R. E. Dessy, R. Kornmann, C. Smith and R. Hayter, J. Chem. Soc., 1966, 88, 5112 Search PubMed.
- R. E. Dessy, F. E. Stary, R. B. King and M. Waldrop, J. Am. Chem. Soc., 1966, 88, 471 CrossRef CAS.
- J. A. McCleverty, N. M. Atherton, J. Locke, E. J. Wharton and C. J. Winscom, J. Am. Chem. Soc., 1967, 89, 6082 CrossRef CAS.
- G. B. Carpenter, G. S. Clark, A. L. Rieger, P. H. Rieger and D. A. Sweigart, J. Chem. Soc. , Dalton Trans, 1994, 2903 RSC.
- If the shift is attributed to the change in the standard redox potential of the reactions 2·Ag(I)+e−→2+Ag (E2·[Ag(I)]) and Ag(I)+e−→Ag (EAg(I)) then the association constant Ag(I)+2=2·[Ag(I)] can be calculated from lnK=nF/RT(EAg(I)
−
E2·[Ag(I)]).
- R. Alberto, W. Nef, A. Smith, T. A. Kaden, M. Neuberger, M. Zehnder, A. Frey, U. Abram and P. A. Schubiger, Inorg. Chem., 1996, 35, 3420 CrossRef CAS.
- S. A. Macgregor, E. J. L. McInnes, R. J. Sorbie and L. J. Yellowlees, Molecular Electrochemistry of Inorganic, Bioinorganic and Organometallic Compounds, eds. A. J. L. Pombeiro and J. A. McCleverty, Kluwer, Dordrecht, 1993, p. 503 Search PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2004 |