DOI:
10.1039/B308308J
(Paper)
New J. Chem., 2004,
28, 91-97
Electrochemical study of Hypocrellin B in acetonitrile and aqueous micellar media. Comparison with Hypocrellin A
Received
(in Montpellier, France)
18th July 2003
, Accepted 11th September 2003
First published on 31st October 2003
Abstract
The electrochemical behaviour of Hypocrellin B (HBH2) has been investigated in acetonitrile and in aqueous micellar media. In organic medium, HBH2 behaves similarly to Hypocrellin A (HAH2) since two successive reversible one-electron transfers are observed in the cathodic region. Exhaustive electrolyses have led to the formation of different reduced forms of HBH2, which have been characterized by spectroscopic investigations. Contrary to HAH2, EPR experiments demonstrated that the one-electron transfer is weakly coupled with protonation phenomenon, leading mainly to the formation of HBH2˙−. Added protons in the medium are required to obtain HBH3˙. In aqueous micellar medium, the first reduction step, whose potential depends on the pH value, is equally observed. This has allowed a diagram of E1/2vs. pH to be established and the determination of the two pKa values of HBH2, whereas the pKa value of the one-electron reduced form of HBH2 has been estimated. The ability of this radical species to produce superoxide anion is discussed and compared with what was previously described for the one-electron reduced forms of HAH2.
Introduction
Since the last decade, numerous works have focused on the study of perhydroxylated polycyclic quinones, which exhibit antitumoral and antiviral activities, including against human immunodeficiency virus.1–5 Hypericin and Hypocrellin A (HAH2) have been thoroughly studied in order to explain the processes that are involved in these activities.2,6–8 It appears that through photochemical irradiation, these photosensitizers induce production of singlet oxygen (1O2, type II mechanism) and the formation of the highly reactive superoxide anion (O2˙−, type I mechanism), which are evoked to explain the phototherapeutic activity of these compounds.9,10 Concerning the type I mechanism, two routes are generally proposed, involving in both cases the reduction of O2 by the one-electron reduced form of the photosensitizer; this reduced form is produced either by the action of the excited state with a donor or by a disproportionation phenomenon of this excited species. In the first case, this can be done since in in vivo conditions, the excited quinone derivative can play the role of oxidant towards numerous organic molecules such as amino acids or lipids, leading to the reduced form of the photosensitizer, which presents highly reducing properties as previously reported.11–13 Recently, a similar compound has been equally studied, Hypocrellin B (HBH2), which differs from the A analogue in the nature of the side chain14,15
(Scheme 1). This natural photosensizing agent, which can be extracted from Skiroia bambusicola, displays antitumoral activity under aerobic conditions, implying either singlet oxygen formation7 or a radical mechanism.16 In this context, as previously examined for HAH2,13 we were interested in the determination of the redox potential of HBH2 in the ground state in order to quantify the reducing properties of the reduced form of this photosensitizer towards molecular oxygen. In this paper, we present the first electrochemical study of HBH2 in both organic and aqueous micellar medium. In organic medium (CH3CN) the redox properties of HBH2 are examined focusing particularly on the stability versus time of its reduced forms and on their protonic behaviour. In aqueous micellar medium, containing Brij 35 in order to reach a convenient concentration (0.1 mM) and to reproduce in vivo conditions, the electrochemical behaviour has been investigated focusing on the influence of the pH on the redox potential of the first reduction step since protonation phenomenon can be coupled to this one-electron transfer as we previously observed for structurally similar compounds.13,17 Combined with this electrochemical study, spectroscopic characterization (UV-visible and EPR) of HBH2 and its reduced forms was effectuated. All these results are compared with those determined for HAH2.13
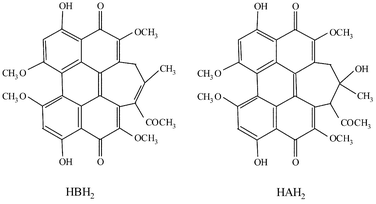 |
| | Scheme 1 Structures of HAH2 and HBH2. | |
Experimental
Cyclic voltammograms (CV), carried out under anaerobic conditions, were recorded using an EGG 273 potentiostat and an EGG 264 A polarographic analyzer for experiments performed in CH3CN and aqueous micellar electrolytes, respectively. For studies in aqueous micellar medium, adsorption phenomena favoured on mercury drop electrodes are likely to be involved in the better resolution of the CV (at 100 mV s−1) compared to results obtained on solid electrodes. Meanwhile, when operating at a lower potential scan rate and taking into account the weak concentration of the photosensitizer, the CV becomes ill-resolved. In CH3CN electrolyte, CV were performed using a vitreous carbon disc (diameter 5 mm) as the working electrode. E1/2 values have been determined by the means of the cathodic and anodic potentials (CV), the same values were obtained using voltamperometry (v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =5 mV s−1) in both organic and aqueous micellar medium. An Ag/Ag+ (10 mM)+CH3CN+0.1 M tetra-n-butylammonium perchlorate (TBAP) electrode and an Ag/AgCl aqueous saturated KCl electrode were used as reference electrodes in CH3CN and aqueous electrolyte, respectively. In order to compare the results in organic and aqueous micellar media, potentials have been converted to the NHE scale using Ag/Ag+ (10 mM)+CH3CN+0.1 M TBAP=0.55 V vs. NHE and Ag/AgCl=0.20 V vs. NHE.18
=5 mV s−1) in both organic and aqueous micellar medium. An Ag/Ag+ (10 mM)+CH3CN+0.1 M tetra-n-butylammonium perchlorate (TBAP) electrode and an Ag/AgCl aqueous saturated KCl electrode were used as reference electrodes in CH3CN and aqueous electrolyte, respectively. In order to compare the results in organic and aqueous micellar media, potentials have been converted to the NHE scale using Ag/Ag+ (10 mM)+CH3CN+0.1 M TBAP=0.55 V vs. NHE and Ag/AgCl=0.20 V vs. NHE.18
Dry TBAP (Fluka), used as the supporting electrolyte in CH3CN medium, was purified as previously described.13 Commercial phosphate salts (Aldrich) were used without any further purification for buffered aqueous micellar solutions between pH=3 and 12. In our experimental conditions no interaction between the quinone derivative and phosphate salt has been displayed, the spectroscopic behaviour of HBH2 not being modified by the addition of such an electrolyte. CH3CN (Rathburn) was used as received, nitrogen flushed and the experiments were carried out under an argon atmosphere in a dry glove box. UV-visible spectra were recorded on an HP 8456 spectrophotometer for experiments performed in CH3CN solution and a Perkin–Elmer apparatus in aqueous micellar medium. Electron paramagnetic resonance (EPR) spectra were recorded with a Brucker ESP 300 E spectrometer at room temperature (22–24°C) with the following parameters: microwave power of 1 mW, modulation amplitude of 0.197 G, time constant of 327.68 ms, scan rate of 1342 s, scan width of 8 G, X-band, modulation frequency of 100 kHz for experiments carried out in acetonitrile. In aqueous micellar medium the same parameters were applied excepted for the scan rate and the scan width, which were equal to 671 s and 13 G, respectively.
Due to the very low solubility of HBH2 in aqueous medium, micellar electrolyte has been used in order to reach a more convenient concentration. Although these solvation conditions do not reflect the parameters of an aqueous medium, they allow in vivo conditions to be reproduced. Brij 35 (Merck) micellar solutions were prepared following the injection method previously described.19 An aqueous solution of Brij 35 (0.1 M) containing HBH2
(0.2 mM) was injected into a buffered aqueous phosphate solution at different pH values (concentration of the phosphate salt was 0.2 M). The final Brij 35 concentration (50 mM) in aqueous micellar solution was above the critical micellar concentration (0.2 mM). The Hypocrellin B concentration in aqueous micellar solution was determined by UV-visible titration. By means of the absorption at λ=464 nm (ε=21000 L mol−1 cm−1 at pH=7), one can establish that close to 0.1 mM HBH2 concentration has been obtained.
The electrolyses in micellar medium have been performed using a vitreous carbon plate as working electrode. It should be noted that, in these conditions, the strong background current prevents a precise determination of the coulombic yield for the one-electron reduction of HBH2. Moreover, addition of Brij 35 in greater excess does not induce modification of the electrochemical behaviour of HBH2. Concerning the spectroscopic investigation in aqueous micellar medium, experiments have been carried out using more dilute HBH2 solutions (i.e., 5 µM). pH measurements have been effectuated by previously calibrating the pH electrode (PHM 210 Metterlab) using several commercial (Aldrich) buffered solutions (pH 4, 5, 6, 7, 9, 12).
Results and discussion
Electrochemical behaviour in acetonitrile medium
As for Hypocrellin A (HAH2)13
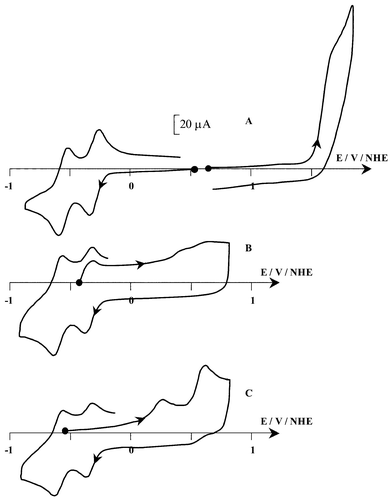
(Scheme 1), the reduction can be accomplished by two successive one-electron steps. The cyclic voltammogram (CV)
(Fig. 1) on a vitreous carbon electrode in CH3CN+0.1 M TBAP exhibits [Fig. 1(A)], in the cathodic region, two well-defined one-electron reversible reductions at E1/2=−0.40 and −0.64 V (ΔEp=60 mV). In the anodic part, one irreversible oxidation is observed at Epa=1.58 V. The comparison of this electrochemical behaviour with that of HAH2 shows that the modification of the lateral chain within the photosensitizer leads to a decrease of the potential value of all the redox processes. This variation is well-observed for the first cathodic process and the anodic one whereas for the second reduction step, this decrease is less pronounced. Concerning the first reduction step, E1/2(HBH2) appears 30 mV more negative compared to E1/2(HAH2). This shift towards a more negative value can be due to the different nature of the lateral chain that, in the case of HBH2, due to the presence of double bond, contributes to an increase of the conjugation network compared to HAH2. Such an influence is equally observed in the anodic part, since, although this process is irreversible, the electron transfer appears at a lower potential than that of HAH2
(1.66 V).
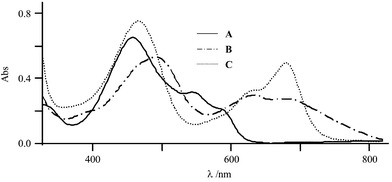
The one-electron reduction of HBH2 is confirmed by exhaustive electrolysis at −0.50 V, which consumes close to one faraday per mole of photosensitizer. The reduction leads to some drastic changes in the visible spectrum as judged by a change in the red colour of the starting solution to a green one. This is illustrated by the strong decrease of the band of the initial compound at 548 nm (ε=10000 L mol−1 cm−1) and the appearance of two new broad bands at 631 and 683 nm (ε=9500 and 8800 L mol−1 cm−1, respectively)
(Fig. 2). This is accompanied by a bathochromic shift of the band at 460 nm to reach 492 nm. One can remark that a shift of the same order of magnitude has been observed during the one-electron reduction of HAH2. Moreover, very similar spectroscopic modifications have been detected in the photoinduced reduction of HBH2 in ethanol buffer at pH=8.5.20
The CV of the resulting solution shows, beside the expected oxidation peak of HBH2˙− at Epa=−0.36 V [Fig. 1(B)], the presence of two weak peaks at more positive potentials (0.44 and 0.58 V). This result demonstrates that HBH2˙− is the major product issued from this reduction and suggests that this one-electron transfer is weakly associated with coupled reactions. It should be noticed that these coupled reactions are reversible on the time scale of the CV since the typical electrochemical behaviour of the starting compound is restored on the reverse scan of the CV. This is confirmed by re-oxidation of the reduced solution (electrolysis at 0.78 V), which leads to the total restitution of the initial amount of HBH2 as judged by CV and visible titration. Given that these side reactions are reversible, one can suggest that proton transfer (by the residual water present in the electrolyte) or homoassociation phenomena could be associated with this reduction step, as previously evoked for HAH2.13 The EPR spectrum (Fig. 3) of the reduced solution shows unambiguously that the reduction leads mainly to one radical species. The hyperfine structure is well-resolved and the determination of the splitting constants (Table 1) are in good agreement with those already determined for HAH2˙−13 and for HBH2˙− prepared in ethanol–buffer solutions through photochemical irradiation in the presence of an electron donor.20 Moreover, the Landé
g factor value for HBH2˙−
(g=2.0047) is the same as that of HAH2˙−, demonstrating that the eventual contribution of proton transfer associated with the electron transfer should be considered as minor. In addition, taking into account that electrochemistry and EPR spectroscopy show that this reduction step leads mainly to HBH2˙−, one can suggest that this anionic species appears to be relatively less basic than HAH2˙− since under the same experimental conditions, electrolysis of Hypocrellin A produces simultaneously two radical species, HAH2˙− and HAH3˙, in a 1∶1 ratio.13
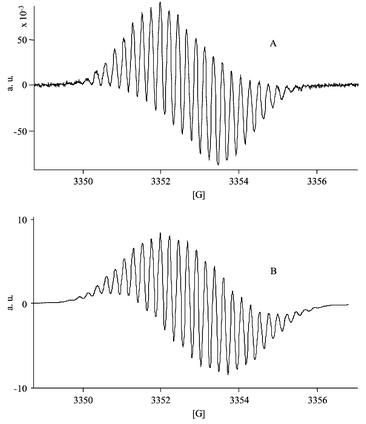 |
| | Fig. 3 (A) EPR spectrum of the one-electron reduced form of HBH2
(0.4 mmol·L−1) obtained by exhaustive electrolysis at −0.50 V in acetonitrile+0.1 M TBAP medium. (B) Simulated spectrum of HBH2˙−. | |
This less basic character of HBH2˙−, compared to HAH2˙−, can be due to the presence of a double bond in the side chain, improving the delocalization of the electrogenerated negative charge. In order to identify the proton interaction with HBH2˙−, electrochemistry has been performed in acidic conditions (i.e., by addition of one or two molar equivalents of perchloric acid). Under these conditions, the visible spectrum HBH2 is unaffected by the added protons. The exhaustive electrolysis of the acidic solution consumes one faraday per mole of compound and leads to a deep green solution. HBH3˙ is characterized by two new bands at 474 and 620 nm (ε=20200 and 15300 L mol−1 cm−1, respectively) and a shoulder at 730 nm (ε=8500 L mol−1 cm−1). Surprisingly, whereas for HAH2 addition of acid in the medium leads to an increase of the g factor value, the EPR analysis of the reduced acidic solution of HBH2 shows that the g factor value for HBH3˙
(g=2.0045) is lower than that of HBH2˙−. Moreover, whereas the EPR spectra of HAH3˙ and HAH2˙− are significantly different, the spectrum of HBH3˙ has the same shape as that of HBH2˙−. The absence of important modifications in the shape of both EPR spectra can be due to the important contribution of the splitting constant of the protons of the lateral chain, which are engaged in the overall conjugation in HBH2
(Table 1). The simulated spectra of HBH3˙ and HBH2˙− are represented in Figs. 3 and 4, showing that the contribution of the side chain of HBH2 induces an high similitude of the spectra.
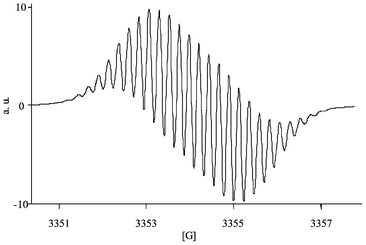 |
| | Fig. 4 Simulated spectrum of HBH3˙. | |
Table 1 Experimental and simulated EPR spectral parameters of the one-electron reduced forms of HBH2
| |
|
Splitting constant |
| Species |
g factora |
Phenolic hydroxyb |
Aromaticb |
Methoxyb |
Side chainb |
Linewidtha/G |
|
Experimental.
Simulated
|
| HBH2˙− |
2.0047 |
1.38 (1H) |
0.47 (1H) |
0.23 (6H) |
0.68 (5H) |
0.3 |
| HBH3˙ |
2.0045 |
0.47 (2H) |
0.7 (2H) |
0.23 (12H) |
0.23 (5H) |
0.3 |
| 1.35 (1H) |
This study has been extended to the second reduction process. The exhaustive electrolysis of a HBH2 solution leads to a green solution (λ=468, 630 and 685 nm; ε=24300, 10500 and 16200 L mol−1 cm−1, respectively; Fig. 2). The CV of the resulting reduced solution now shows two new irreversible peaks at 0.26 and 0.60 V, whereas on the subsequent cathodic scan the typical HBH2/HBH2˙− and HBH2˙−/HBH22− redox systems are almost entirely recovered. Moreover, the initial amount of the starting compound can be restored by exhaustive oxidation at 0.88 V, consuming two faradays per mole. As for HAH213 and Hypericin,17 one can suggest that, given the strong basic character of the dianionic reduced form of the photosensitizer, protonation/deprotonation phenomena should be taken into account in this overall redox process.
Chemical behaviour in aqueous micellar medium
We observed that the protonation step could be coupled to the electron transfer; thus, since HBH2 bears labile protons, the first step of this study consists in the determination of the acid-base properties of this photosensitizer in aqueous micellar medium.
Determination of pKa values for HBH2.
Two acid-base couples can be involved (i.e., HBH2/HBH− and HBH−/HB2−), which can be clearly quantified by visible spectroscopy. Two drastic changes vs. pH are observed (Fig. 5). In the pH range 4.0–10.0, the first pKa value can be estimated from the decrease of the band at 548 nm (ε=10000 L mol−1 cm−1 at pH=7) concomitantly with the bathochromic shift of the absorption band from 460 nm (ε=21600 L mol−1 cm−1) up to 477 nm (ε=16000 L mol−1 cm−1). For higher pH values, the second pKa value is determined from the red shift of the absorption band from 638 nm to 643 nm and to a smaller extent by the increase of absorption at 638 nm. Similar spectroscopic modifications have been observed for the two deprotonation steps of HAH2.13 Thus, pKa values have been determined for eqns. (1) and (2):| | | HBH−⇆HB2−+H+ pKa2=11.7 | (2) |
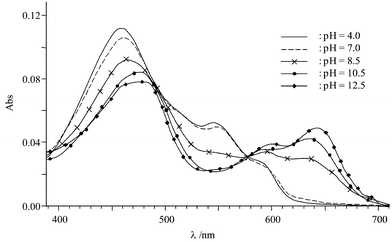 |
| | Fig. 5 Absorption spectrum vs. pH of Hypocrellin B (5 µmol·L−1) in buffered aqueous micellar solution. | |
These two successive deprotonation steps can also be observed by evolution of the luminescence spectra vs. pH. By photoexcitation at 460 or 548 nm in acidic medium (in the pH range 4–7), an intense fluorescence band characteristic of HBH2 is observed at 612 nm, which in more basic conditions strongly decreases until it disappears entirely at pH=10.5 (Fig. 6). For higher pH values, a new emission band at 668 nm is observed. In these experimental conditions, the same pKa values for the two acid-base couples have been obtained. The comparison of these values with those determined for HAH2
(i.e., 8.2 and 11.4)13 shows here that the chemical modification of the lateral chain has a minor influence on the acidic properties of HBH2. Moreover, these values are in good agreement with those determined by He et al.20 in hydroethanolic solution (pKa=8.4 and 11) showing that solvation conditions have only a slight effect on the first deprotonation step. On the other hand, a more significant effect is observed concerning the second pKa value.
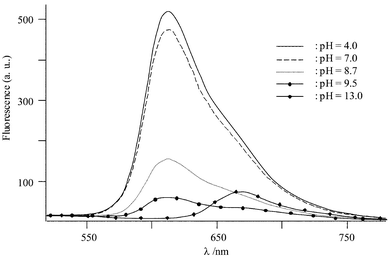 |
| | Fig. 6 Emission spectrum vs. pH of HBH2
(5 µmol·L−1) in buffered aqueous micellar solution. | |
Electrochemical behaviour of HBH2 in aqueous micellar medium
As previously observed for HAH2, the shape of the CV of HBH2 in aqueous micellar medium depends on the acidic character of the solution. In acidic conditions (pH<7), one reversible redox reduction process is observed whose E1/2 value depends on the pH of the medium (Fig. 7), varying from E1/2=0.085 V at pH=3.4 to reach E1/2=−0.092 V at pH=6.8 (i.e., 51 mV per pH unit). This pH dependency, which exhibits a slope relatively close to the theoretical one for a one-electron process (i.e., 60 mV per pH unit), as previously observed for HAH2, demonstrates that, in this pH range, the one-electron reduction is coupled to proton transfer. Taking into account that in these experimental conditions HBH2 is not deprotonated, one can suggest that this process is relative to the redox couple HBH2/HBH3˙ [eqn. (3)]:It should be noted that in this pH range ΔEp values (≈30 mV) are lower than the theoretical value (60 mV). This cannot be attributed to a redox process involving a bielectronic transfer since, when using vitreous carbon as the working electrode, the ΔEp values increase to reach 70 mV, although the CV becomes less resolved due to the important contribution of the background current. The same electrochemical behaviour was observed for HAH2. Moreover, the reduced solutions (by exhaustive electrolyses) present, in all cases in the pH range 4–9, EPR spectra attesting to the monoelectronic character of this electrochemical process. On the other hand, as previously proposed,20–22 bielectronic reduction can be evoked in more drastic conditions. In basic medium (pH>11.7), since the predominant form is HB2−, the absence of intramolecular hydrogen bonds contributes to a decrease in the stability of the radical species and accelerates the formation of the two-electron reduced hydroquinone. Moreover, in acidic conditions (below pH=4), the hydroquinone derivative can be equally produced due to disproportionation of HBH3˙, which may be catalysed by protons. It should be noted that, depending on the solvent (acetonitrile or aqueous micellar media), the E1/2 values appear significantly different (−0.40 and −0.092 V at pH=6.8, respectively). This discrepancy could be assigned to homoassociation phenomenon favoured in aqueous micellar medium, which leads to the formation of the more easily reducible dimer 23 as previously reported for HAH2.13
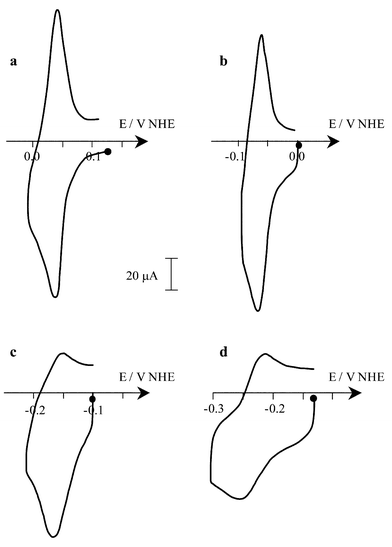 |
| | Fig. 7 Cyclic voltammograms (v=100 mV s−1) on a mercury electrode of 0.1 mM HBH2 in buffered aqueous micellar solution+phosphate (0.1 M) at different pH: (a) 4.3, (b) 6.4, (c) 8.5, (d) 10.5. | |
In more basic conditions (6.8<pH<8.3), the E1/2 value remains constant at ca.
−0.094 V (Fig. 7). Thus, in this pH range, a redox process precluding a protonation step has to be considered [eqn. (4)]:
allowing the determination of the p
Ka of the HBH
3˙/HBH
2˙
− couple [
eqn. (5)]:
| | | HBH2˙−+H+⇆HBH3˙ pKa3=6.8 | (5) |
Thus, considering that the error in the determination of the p
Ka3 value is ±0.1 pH unity, one can suggest that HBH
2˙
− appears as a relative less basic species compared to HAH
2˙
−
(p
Ka=
7) as proposed before in the electrochemical study of HBH
2 in
acetonitrile medium.
For pH>8.3, since HBH− is predominant in these experimental conditions, the E1/2vs. pH variation from −0.115 V at pH=8.5 up to −0.235 V at pH=11.0 (i.e., 50 mV per pH unit) is relative to the redox couple HBH−/HBH2˙− [eqn. (6)]:
Under higher pH conditions, the CV becomes ill-resolved, precluding an accurate determination of the
E1/2 value. In addition, up to pH 11 we do not observe a plateau corresponding to the HBH
−/HBH˙
2− redox couple as detected for HAH
2, demonstrating that deprotonation of HBH
2˙
− occurs in stronger basic conditions compared to HAH
2
(p
Ka HAH
2˙
−/HAH˙
2−=
8.5). All these results are illustrated by the
E1/2vs. pH diagram (
Fig. 8).
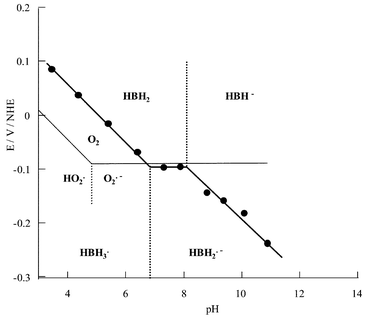 |
| | Fig. 8
E
1/2
vs. pH diagram of HBH2 in buffered aqueous micellar solution. | |
Attempts to correlate this diagram with the variation of the g factor value vs. pH were unsuccessful. Although EPR spectra were obtained over a large pH range, the poor resolution and the high similitude of the spectra of HBH3˙ and HBH2˙− do not facilitate the precise determination of g factor values. This E1/2vs. pH diagram allows a better understanding of the production of superoxide radical through photochemical irradiation of HBH2. O2˙− production can be mainly achieved by several processes involving the excited form of HBH2 [eqns. (7)–(9)]:
| | | (a) HBH2*+HBH2*⇆HBH2˙−+HBH2˙+ | (7) |
| | | (a) HBH2*+D⇆HBH2˙−+D˙+ | (8) |
Although singlet oxygen production should not be forgotten, this contribution can be considered as minor in micellar aqueous medium, as recently observed by Yu
et al.14Eqns. (7) and (8) involve the transient formation of HBH
2˙
− whereas in
eqn. (9), superoxide anion is produced by interaction between molecular oxygen and the excited state of the photosensitizer. In each process, the excited state of the photosensitizer (HBH
2*) is involved.
In our experimental conditions, as for HAH2, the triplet state of HBH2* also has to be considered since its lifetime (12.5 µs) is longer than that of the singlet state (1.2 ns). Moreover, in this medium, the quantum yield of formation of this triplet state is rather high (0.77).
In eqn. (7), a high concentration of photosensitizer is required, since this mechanism requires the reaction between two excited molecules. This can be reached considering a high local concentration of the photosensitizer, such as that within a liposome or micelle. Eqn. (8) involves the reaction of HBH2* and a donor. Using the Rehm–Weller relation,24 from the energy of the triplet state (1.74 eV7), the redox potential of the couple HBH2*/HBH2˙− can be estimated to be equal to 1.65 V vs. NHE (at pH=7). Thus, in the presence of a donor with a redox potential lower than 1.65 V, such as amino acids or proteins, HBH2˙− can be thermally produced.
Comparison of the E1/2 values with the one relative to the O2/O2˙− redox system (including the protonation of O2˙− with E°=−0.090 V, pKa O2˙−/HO2˙=4.825), one can establish that in our experimental conditions, superoxide anion formation is observed for pHs higher than 6.7 whereas for HAH2, this pH value has been estimated to be equal to 6.4.13 Thus, although the contribution of the side chain in HBH2 leads to the slight increase of the potential of the first one-electron reduction, it leads to an important modification of the redox properties of the reduced form of the photosensitizers towards molecular oxygen. Moreover, since in strong basic conditions (pH>11) the reduced form of HBH2 seems to be rather unstable,22 HBH2˙− appears to be the single species able to reduce molecular oxygen to O2˙−. The comparison of the electrochemical behaviour of HAH2 and HBH2, shows that in close to neutral conditions (pH 6.8–8.3) O2˙− formation should be thermodynamically more favoured for HAH2 [E1/2(HAH2)=−0.110 V,13E1/2(HBH2)=−0.092 V, E° O2/O2˙−=−0.090 V] since ΔE
(=E° O2/O2˙−−E1/2) is close to zero for HBH2 whereas this value is more positive for HAH2. On the other hand, in more basic conditions (8.3<pH<10) HBH2˙− presents a more important reducing character compared to the reduced form of HAH2. Indeed, the ΔE value drastically decreases vs. pH for HBH2, to reach 0.10 V when for HAH2 this value remains constant (0.03 V).
For eqn. (9), the estimation of the contribution of this equilibrium requires a knowledge of the E1/2 value of the HBH2*/HBH2˙+ redox system, which cannot be determined since, in aqueous micellar medium, the oxidation of the solvent occurs at a low potential, precluding the observation of the anodic process relative to the photosensitizer. Although following an analogous phenomenon, we previously demonstrated in a recent work,26 that, in acetonitrile electrolyte, photochemical production of HBH2˙+ and O2˙− is thermally unfavoured, in aqueous micellar medium, this redox step could be accomplished taken into account that the nature of the solvent has an important influence on the potential values of the redox systems involved.
Thus, given that singlet oxygen formation does not occur14 and that O2 reduction by photoinduced HBH2˙− appears to be thermally poorly efficient in physiological conditions (pH around 7), one can suggest that in our experimental conditions, superoxide anion production through HBH2* has to also be considered.
Conclusion
The electrochemical behaviour of HBH2 has been investigated in acetonitrile and aqueous micellar media. In both solvation conditions, the first one-electron reduction is reversible on the time scale of the CV. In organic medium, exhaustive reduction leads mainly to HBH2˙−, showing that this reduced species presents a less basic character compared to HAH2˙−. This discrepancy between the two photosensitizers can be due to the presence of a conjugated side chain in HBH2, allowing a better delocalization of the radical charge. On the other hand, in the presence of added proton, HBH3˙ can be obtained and characterized. In aqueous micellar medium, a similar behaviour is observed, since in acidic conditions HBH3˙ is obtained whereas, for pH between 6.8–11, the reduction leads to HBH2˙−, confirming the lesser ability of HBH2˙− to be protonated compared to HAH2˙−. This study has allowed the determination of the pKa value of the HBH3˙/HBH2˙− couple and the confirmation of those of HBH2/HBH− and HBH−/HB2−.
This electrochemical behaviour has important mechanistic implications since we demonstrated that among the different one-electron reduced forms of HBH2, HBH2˙− should be the only species that is able to reduce molecular oxygen, superoxide anion formation being exothermic for pH higher than 6.7. The comparison of the electrochemical behaviour of HAH2 and HBH2 in aqueous micellar medium has allowed comparison of their reducing properties towards O2. In close to biological conditions, O2˙− production appears to be thermally more favoured for HAH2, whereas, in more basic conditions, superoxide anion formation is more efficient using HBH2. Meanwhile, the formation of O2˙− by a bimolecular process involving HBH2* and O2 should be also considered.
Acknowledgements
We are grateful to Dr. A. Deronzier (LEOPR) for fruitful discussions.
References
- H. Falk, Angew. Chem., Int. Ed., 1999, 38, 3117 CrossRef CAS.
- R. M. Gulick, V. McAuliffe, J. Holden-Wiltse, C. Crumpaker, L. Liebes, D. S. Stein, P. Meehan, S. Hussey, J. Forcht and F. T. Valentine, Ann. Inter. Med., 1999, 130, 510 Search PubMed.
- J. B. Hudson, J. Zhou, J. Chen, L. Harris, L. Yip and G. H. Towers, Photochem. Photobiol., 1994, 60, 253 CAS.
- J. Park, D. S. English, Y. Wannemuhler, S. Carpenter and J. W. Petrich, Photochem. Photobiol., 1998, 68, 593 CAS.
- S. M. Ali, S. K. Chee, G. Y. Yan and M. Olivo, J. Photochem. Photobiol. B, 2001, 65, 59 CrossRef CAS.
- H. Bouirig, D. Eloy and P. Jardon, J. Chim. Phys., 1992, 89, 1391 CAS.
- Z. Diwu and J. W. Lown, J. Photochem. Photobiol. A, 1992, 64, 273 CrossRef CAS.
- J. B. Hudson, V. Imperial, R. P. Haugland and Z. Diwu, Photochem. Photobiol., 1997, 65, 352 CAS.
- Z. Diwu and J. W. Lown, J. Photochem. Photobiol. B, 1993, 18, 131 CrossRef CAS.
- C. Hadjur and P. Jardon, J. Photochem. Photobiol. B, 1995, 29, 147 CrossRef CAS.
- J. Redepenning and N. Tao, Photochem. Photobiol., 1993, 58, 532 CAS.
- L. Y. Zhang, B. R. Misra and H. P. Misra, Photochem. Photobiol., 1990, 52, 677.
- S. Dumas, P. Jardon, J. C. Lepretre and A. Jeunet, New J. Chem., 2001, 25, 1313 RSC.
- C. Yu, S. Xu, S. Chen, M. Zhang and T. Shen, J. Photochem. Photobiol. B, 2002, 68, 73 CrossRef CAS.
- E. P. Estey, K. Brown, Z. Diwu, J. Liu, J. W. Lown, G. C. Miller, J. Tulip and M. S. McPhee, Cancer Chemother. Pharmacol., 1996, 37, 343 Search PubMed.
- Z. Diwu, Photochem. Photobiol., 1995, 61, 529 CAS.
- L. Burel, P. Jardon and J. C. Lepretre, New J. Chem., 1997, 21, 399 Search PubMed.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97 CrossRef.
- D. Eloy, A. Lepellec and P. Jardon, J. Chim. Phys., 1996, 93, 442 CAS.
- Y. Y. He, J. Y. An and L. J. Jiang, Dyes Pigm., 1999, 41, 79 CrossRef CAS.
- Y. Z. Hu, J. Y. An and L. J. Jiang, Acta Chim. Sin., 1992, 50, 186 Search PubMed.
- K. Reska and J. W. Lown, Photochem. Photobiol., 1989, 50, 297 CAS.
- Z. Diwu, L. J. Jiang and N. H. Zhang, Sci. China, 1990, 33, 18 Search PubMed.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259 CAS.
-
G. R. Buettner and B. Jurkiewicz, in Analysis of Free Radicals in Biological Systems, eds. A. E. Favier, J. Cadet, B. Kalyanaraman and J. L. Pierre, Birkhaüser, Basel, 1997, p. 147 Search PubMed.
- S. Dumas, J.-C. Lepretre, A. Lepellec, A. Darmanyan and P. Jardon, J. Photochem. Photobiol. A, 2003 Search PubMed
(in
press).
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2004 |
Click here to see how this site uses Cookies. View our privacy policy here.