Evidence for the in situ formation of copper acetylides during Pd/Cu catalyzed synthesis of enynes: a new synthesis of allenynols†
Philippe
Bertus
a,
Fabien
Fécourt
b,
Claude
Bauder
b and
Patrick
Pale
*b
aLaboratoire des Réactions Sélectives et Applications (CNRS UMR 6519), Université de Reims-Champagne-Ardenne, BP 1039, 51687, Reims, France
bLaboratoire de Synthèse et Réactivité Organique (CNRS UMR 7123), Institut Le Bel, Université L. Pasteur, 4 rue B. Pascal, 67000, Strasbourg, France. E-mail: ppale@chimie.u-strasbg.fr; Fax: +33 3 9024 1517; Tel: +33 3 9024 1517
First published on 28th November 2003
Abstract
The in situ formation of copper acetylide in Pd/Cu catalyzed coupling reactions of acetylenic derivatives has been demonstrated by the reaction with ethynyloxiranes, which provides allenynols; a new Cu catalyzed route to the latter has also been found.
Pd/Cu catalyzed cross-coupling reactions between terminal acetylenes and aryl or vinyl halides or triflates, the so-called Sonogashira reaction, have been recognized since the mid-seventies.1,2 The mildness and the efficiency of these methods have since led to an impressive collection of applications in numerous areas from medicinal chemistry to material science.3,4 Despite being widely utilized, the exact mechanism of this C–C bond formation has so far not been demonstrated. Although the main features of the Pd catalytic cycle have been established,5 the Cu catalytic cycle is still poorly understood.6 In this communication, we present chemical evidence for the in situ formation of copper acetylides, which allow us to propose a mechanism for the Cu catalytic cycle of Pd/Cu catalyzed cross-coupling reactions.
Several research programs dedicated to the synthesis of polyunsaturated natural products led us to study the formation of epoxyenynes by coupling reactions7,8 and to begin the investigation of the mechanism of such reactions.9 The direct synthesis of epoxyenynes starting from ethynyloxiranes and vinyl triflates or halides proved to be inefficient with the reported procedures.7a,10 In every trial,7 the ethynyloxirane engaged was rapidly consumed while only a small amount of the expected enyne was obtained (less than 25%, see Scheme 1) among various other polar side-products.
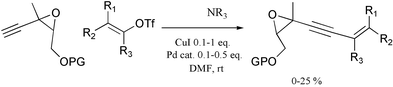 | ||
| Scheme 1 Inefficient cross-coupling of ethynyloxiranes to vinyl triflates. | ||
Even when the ethynyloxirane was slowly added to the other components of the reaction, decomposition was still the major process; the yield of enyne was nevertheless better but still modest (40–50%). Whatever the conditions, reaction monitoring indicated that the triflate component was almost unchanged while the acetylenic epoxide was rapidly consumed.
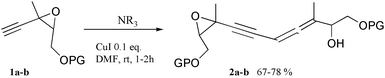
To get some understanding on the evolution of epoxyacetylenes during Sonogashira coupling, we tested a model acetylenic epoxide (1a) with each reagent (palladium complexes, amines and copper salts) and selected combinations of these, depending on the expected reactivity (Table 1). The epoxide 1a was obtained by meta-chloroperbenzoic acid epoxidation of the commercially available Z 3-methylpent-2-en-4-yn-1-ol and protection as a tert-butyldiphenylsilyl ether.
| Ethynyloxirane | Conditions | Time | Yielda | Product | |
|---|---|---|---|---|---|
| a Yields of isolated pure products. b Increasing amounts from 0.1 to 0.5 equiv. have been used. c Various amines (diethyl-, triethyl- and ethyldiisopropylamine) have been used with similar results. d Increasing amounts from 0.1 to 1 equiv. have been used. e Results obtained with 0.1 equiv. of CuI. | |||||
| 1 |
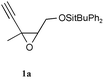
|
Pd(PPh3)4b | 2 days | 0 | — (1a recovered) |
| 2 | Pd(OAc)2(PPh3)2 | 2 days | 0 | ||
| 3 | NR3c | 2 days | 0 | ||
| 4 | Pd(PPh3)4, NR3c | 2 days | 0 | ||
| 5 | Pd(OAc)2(PPh3)2, NR3c | 2 days | 0 | ||
| 6 | CuId | 2 days | Traces |
|
|
| 7 | CuI,e HNEt2 | 2 h | 78% | ||
| 8 | CuI,e NEt3 | 0.7 h | 67% | ||
| 9 | CuI,e EtNiPr2 | 1 h | 72% | ||
| 10 |
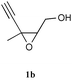
|
CuI,e HNEt2 | 1 h | 69% |
|
In the presence of zero- or divalent palladium complexes in DMF, no transformation of the acetylenic epoxide 1a was observed (entries 1 and 2) at room temperature, even when an amine was added (entries 4 and 5). These results are surprising since palladium complexes are known to catalyze the selective opening of related vinyloxiranes, especially in the presence of certain amines.11 Although amines are known to selectively open acetylenic epoxides,12 amines alone in DMF added to 1a did not afford any significant transformation at room temperature (entry 3). In the presence of copper iodide alone, only a slow evolution was observed (entry 6). However, when copper iodide and an amine were added, a rapid evolution occurred and a new polar product was formed along with only a few polar side-products (entries 7–9). This major product was isolated and characterized, mainly by spectroscopy.
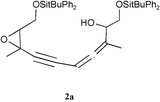
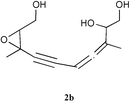
The 1H NMR spectrum revealed the presence of the tert-butyldiphenylsilyl group. However, the signal intensity of the aromatic protons relative to the intensity of the other protons suggested that two silyl groups are in fact present in this molecule. In this spectrum, the presence of the typical signals of the starting 2,3-epoxypent-4-ynol, a triplet at 3.1 ppm coupled to an ABX system at 3.80–3.95 ppm, clearly showed that a fragment identical to the starting epoxyalkyne was preserved in the structure of the new product. Therefore, the new product may result from the condensation of two molecules of the starting silylated epoxyalkynol. The IR spectrum indeed exhibited a signal corresponding to a disubstituted triple bond (ν![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =2235 cm−1). However, this spectrum also showed the presence of new bands, one at 3400 cm−1 typical of a hydroxyl function, and another one at 1960 cm−1 suggesting the presence of an allene group. Further NMR analysis confirmed this attribution. The 13C NMR spectrum exhibited one signal at 210 ppm and two others located at around 76 ppm, which can indeed be attributed to an allene. HMQC experiments showed that the carbon signals at 210 and 77 ppm do not correlate with any proton but the third signal at 76 ppm correlates with a multiplet at 5.25 ppm corresponding to one proton. Others correlations also support this attribution and allowed us to propose the epoxyallenynol structure 2a for this compound (Scheme 2). The splitting of some NMR signals was clearly indicative of the presence of diastereoisomers.
=2235 cm−1). However, this spectrum also showed the presence of new bands, one at 3400 cm−1 typical of a hydroxyl function, and another one at 1960 cm−1 suggesting the presence of an allene group. Further NMR analysis confirmed this attribution. The 13C NMR spectrum exhibited one signal at 210 ppm and two others located at around 76 ppm, which can indeed be attributed to an allene. HMQC experiments showed that the carbon signals at 210 and 77 ppm do not correlate with any proton but the third signal at 76 ppm correlates with a multiplet at 5.25 ppm corresponding to one proton. Others correlations also support this attribution and allowed us to propose the epoxyallenynol structure 2a for this compound (Scheme 2). The splitting of some NMR signals was clearly indicative of the presence of diastereoisomers.
 | ||
| Scheme 2 Cu catalyzed condensation of ethynyloxiranes. | ||
The corresponding non-protected derivative 1b reacted under the same conditions, giving the corresponding unprotected epoxyallenynol 2b with a slightly lower yield (entry 10). 2b exhibited spectroscopic properties very similar to 2a.
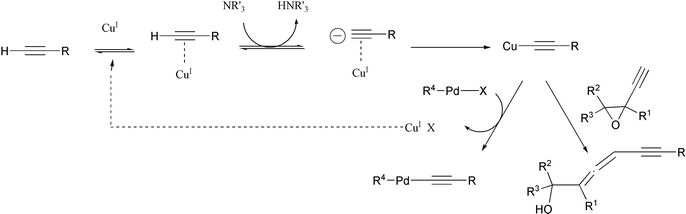
The epoxyallenynol structures determined for 2a and 2b clearly showed that 1a or 1b added to itself through a SN′2 reaction. Since 2a,b were only formed in the presence of copper iodide and amine, it becomes obvious that a copper epoxyacetylide was formed in situ under these conditions. The copper(I) salt probably acted as a Lewis acid and coordinated to the triple bond. This coordination should alter the electronic density over the acetylenic C–H bond, rendering it more acidic. The acetylenic proton could thus be deprotonated even by simple amines, leading to a copper acetylide (Scheme 3, first line). Copper acetylides can indeed be obtained by the treatment of alkynes with an ammoniacal solution of copper chloride.13
 | ||
| Scheme 3 Proposed mechanisms for the Sonogashira coupling reaction and for the presented reaction of ethynyloxiranes. | ||
Starting from ethynyloxiranes, the copper acetylide formed in situ is nucleophilic enough to react with the starting material, leading to allenynol self-adducts (Scheme 3, bottom right). SN′2 reactions of organocopper derivatives to ethynyloxiranes already have precedents in the literature. However, they always refer to stoechiometric processes with pre-formed organometallics.14,15 The reaction described here is thus, to our knowledge, the first catalytic SN′2 reaction of ethynyloxiranes that does not involve pre-formed organometallics.
Since the same allenynol 2a has been detected in our attempts to couple 1a with vinyl triflates in various Pd/Cu coupling conditions (Scheme 1),7 it is now clear that copper acetylides are also formed in situ in these reactions. In Sonogashira cross-coupling reactions, this copper acetylide reacts with the organopalladium species resulting from oxidative addition of a zero-valent palladium complex to vinyl or aryl halide or triflate. This transmetallation step produces a diorganopalladium species and liberates a copper ion, ready for another cycle (Scheme 3, bottom left). However, during the coupling of ethynyloxiranes, the in situ formed copper acetylide is too reactive and it is consumed together with a second ethynyloxirane to form allenynol, to the detriment of coupling products.
The present results demonstrate the in situ formation of copper acetylide in Pd/Cu catalyzed coupling reactions with acetylenic derivatives. They also offer the first catalytic route to allenynols without resorting to pre-formed organometallics.
Work is now in progress in our group to clarify the stereoselectivity of this self-addition16 and to determine the scope and limitations of this new reaction.
Experimental
Typical procedure
To a solution of alkyne (0.6 mmol., 1 equiv.) in DMF (10 mL) were successively added diethylamine (1.2 mmol., 2 equiv.) and copper iodide (11 mg, 0.06 mmol., 0.1 equiv.). The initially slightly yellow solution gradually darkened. After disappearance of the starting material as judged from TLC (2 h), diethyl ether (10 mL) and then a saturated aqueous solution of ammonium chloride (10 mL) were successively added to the reaction mixture. After extraction with diethyl ether, the combined organic layers were washed with water. After drying with sodium sulfate, filtration and solvent evaporation, a yellowish oil was obtained. Flash chromatography (petroleum ether–ethyl acetate 9 : 1) provided pure product as a colorless oil (see Table 1).=3443, 2235, 1962 cm−1. 1H NMR (250 MHz, CDCl3): δ 1.05 (18H, s, tBuSi), 1.45–1.49 (3H, m, H8′), 1.62–1.67 (3H, m, H3′), 2.51–2.56 (1H, m, OH), 3.11 (1H, t, J=5.2, H9), 3.59–3.78 (2H, m, H1), 3.80–3.95 (2H, m, H10), 4.06–4.15 (1H, m, H2), 5.20–5.28 (1H, m, H5), 7.31–7.42 (12H, m, Ar), 7.60–7.70 (8H, m, Ar). 13C NMR (62 MHz - CDCl3): δ 14.55 and 14.67 (C3′), 19.21 (tBuSi), 23.20 (C8′), 26.76 (tBuSi), 52.27 (C8), 63.76 and 64.63 (C2 and C9), 65.88 (C10), 72.03 and 72.11 (C2), 76.07 and 76.36 (C5), 77.71 (C3), 86.70 (C6), 102.52 and 102.82 (C7), 127.66, 127.77, 129.65, 129.84, 132.94, 133.26, 133.47 and 135.51 (Ar), 209.75 (C4). HRMS (EI): m/z 700.3394 (700.3404 calcd for C44H52O4Si2).
Acknowledgements
P. B. thanks the “Ministère de l'Education Nationale, de l'Enseignement Supérieur et de la Recherche” for a doctoral fellowship. P. P. thanks the Institut Universitaire de France for financial support.References
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 4467 CrossRef CAS.
- (a) V. Ratovelomanana and G. Linstrumelle, Tetrahedron Lett., 1981, 22, 315 CrossRef CAS; (b) V. Ratovelomanana and G. Linstrumelle, Tetrahedron Lett., 1984, 25, 6001 CrossRef CAS; (c) M. Alami, F. Ferri and G. Linstrumelle, Tetrahedron Lett., 1993, 34, 6403 CrossRef CAS.
- K. Sonogashira, in Comprehensive Organic Synthesis, eds. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 3, pp. 521–549 Search PubMed.
- K. Sonogashira, in Comprehensive Functional Group Transformations, eds. A. Katritzky and C. Rees, Pergamon, Oxford, 1995, vol. 1, pp. 995 Search PubMed.
- (a) R. Crabtree, The Organometallic Chemistry of the Transition Metals, Wiley, New York, 1988 Search PubMed; (b) For a more detailed analysis, see: C. Amatore, A. Jutand and A. Suarez, J. Am. Chem. Soc., 1993, 115, 9531 Search PubMed; C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314 CrossRef CAS.
- (a) I. E. Campbell, in Organocopper Reagents, A Practical Approach, ed. R. J. K. Taylor, Oxford University Press, Oxford, 1994, pp. 217–235 Search PubMed; (b) L. Hegedus, Transition Metals in the Synthesis of Complex Organic Molecules, 2nd edn., University Science Books, Sausalito, CA, USA, 1999, p. 91 Search PubMed.
- (a) P. Bertus and P. Pale, Tetrahedron Lett., 1996, 37, 2019 CrossRef CAS; (b) P. Bertus and P. Pale, Tetrahedron Lett., 1997, 38, 8193 CrossRef CAS; (c) P. Bertus and P. Pale, J. Organomet. Chem., 1998, 567, 173 CrossRef CAS.
- (a) P. Bertus, U. Halbes and P. Pale, Eur. J. Org. Chem., 2001, 4391 CrossRef CAS; (b) U. Halbes, P. Bertus and P. Pale, Tetrahedron Lett., 2001, 42, 8641 CrossRef CAS; (c) U. Halbes and P. Pale, Tetrahedron Lett., 2002, 43, 2039 CrossRef CAS.
- P. Bertus, S. Dillinger and P. Pale, Org. Lett., 2001, 3, 1661 CrossRef.
- R. S. Reddy, S. Igushi, S. Kobayashi and M. Hirama, Tetrahedron Lett., 1996, 37, 9335 CrossRef CAS.
- (a) B. M. Trost, G. H. Kuo and T. Benneche, J. Am. Chem. Soc., 1988, 110, 621 CrossRef CAS; (b) G. Jähne, A. Müller, H. Kroha, M. Rösner, O. Holzhäuser, C. Meischsner, M. Helsberg, I. Winkler and G. Riess, Tetrahedron Lett., 1992, 33, 5335 CrossRef.
- (a) M. S. Malinovskii, N. G. Krivosheeva, M. P. Khmel, A. G. Yudasina and V. G. Dryuk, Ukr. Khim. Zh., 1973, 39, 1257 Search PubMed; (b) F. Y. Perveev, L. N. Shil'nikova and R. Y. Irgal, Zh. Org. Khim., 1969, 5, 1373; (c) H. Galons, J. F. Girardeau, C. Combet-Farnoux, M. Miocque, C. Dupont and J. Wepierre, Eur. J. Med. Chem., 1979, 14, 165 CAS.
- J. F. Normant, Synthesis, 1972, 70.
- Two competitive mechanisms may contribute to SN′2 substitutions, leading to stereoisomers. See: A. Alexakis, I. Marek, P. Mangeney and J. F. Normant, Tetrahedron, 1991, 47, 1677 Search PubMed.
- (a) P. R. Ortiz de Montellano, J. Chem. Soc., Chem. Commun., 1973, 710 RSC; (b) P. Vermeer, J. Meijer, C. De Graaf and H. Schreurs, Rec. Trav. Chim. Pays-Bas, 1974, 94, 46; (c) R. Epsztein and N. Le Goff, J. Chem. Soc., Chem. Commun., 1977, 679 RSC; (d) C. Cahiez, A. Alexakis and J. F. Normant, Synthesis, 1978, 528 CrossRef CAS; (e) A. Doutheau, A. Saba and J. Gore, Tetrahedron Lett., 1982, 23, 2461 CrossRef CAS; (f) N. Krause, A. Hoffmann-Röder and J. Canisius, Synthesis, 2002, 1759 CrossRef CAS.
- Depending on the exact mechanism, diastereoisomers can be produced. See ref. 14.
Footnote |
| † The authors dedicate this work to the late Prof. J. Chuche, Université de Reims-Champagne-Ardenne, France. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2004 |