Zn2+ catalysed hydrolysis of β-lactams: experimental and theoretical studies on the influence of the β-lactam structure†
Natalia
Díaz
a,
Tomás L.
Sordo
a,
Dimas
Suárez
*a,
Rosa
Méndez
b and
Javier
Martín-Villacorta
*b
aDepartamento de Química Física y Analítica, Universidad de Oviedo, C/Julian Clavería, 8, 33006, Oviedo, Spain. E-mail: dimas@correo.uniovi.es; Fax: +34 985 10 31 25; Tel: +34 985102266
bDepartamento de Química Física y Expresión Gráfica, Universidad de León, 24071, León, Spain
First published on 22nd October 2003
Abstract
We present both experimental and theoretical results on simple model systems of zinc-β-lactamases. Kinetic studies show that the rate of degradation of β-lactam antibiotics in the presence of zinc ions and tris(hydroxymethyl)aminomethane buffers depends markedly on the structure of the β-lactam. Carbapenems are highly reactive whereas monobactam antibiotics like aztreonam, which are known to be non-susceptible to the catalytic action of the metallo-β-lactamases, are less reactive by three orders of magnitude. To complement the experimental studies, density functional calculations were carried out on model systems. These calculations allowed us to characterise the reactive mode of binding between the β-lactam nucleus and Zn2+ ions as well as to rationalise the kinetic trends observed experimentally. Docking analyses are reported for the complex formed between aztreonam and the mononuclear metallo-β-lactamase from Bacillus cereus. On the basis of all the results, we hypothesise that the aztreonam–metallo-β-lactamase complex might be poorly reactive due to a potential interaction of the N-sulfonate group of aztreonam with the essential Zn ion at the active site.
Introduction
Zinc-β-lactamases synthesised by several pathogenic bacteria have recently become a major research and clinical problem1–3 given that these enzymes, in which zinc ion(s) are crucial for catalysis, efficiently hydrolyze nearly all β-lactams, including the versatile broad-spectrum antibacterial carbapenem derivatives (e.g., imipenem) and the most widely used inhibitors of serine β-lactamases (e.g., clavulanic acid).4 There is, however, an important exception; monobactams (e.g., aztreonam), bearing a strongly acidic sulfonic group at the N-1 position, are scarcely susceptible to the action of the zinc-β-lactamases.3,5The metal-ion-catalysed hydrolysis of β-lactams in aqueous solution has gained renewed interest due to its importance as a reference reaction for the zinc-β-lactamases. Thus, the effects of divalent metal ions (Cu2+, Cd2+, Zn2+) on the aqueous hydrolysis of penicillins and cephalosporins have been investigated experimentally to show that metal ions can dramatically enhance the rate of hydrolysis by a factor of 104–107.6 On the basis of the saturation appearance of the kobsvs. [M2+] plots and the linear dependence of log kobsvs. pH, it has been proposed that the metal ions bind to the β-lactam carboxylate group, promoting the attack of an external hydroxide on the β-lactam carbonyl group. The same appears to be true for the Zn2+-promoted methanolysis of nitrocefin.7 This proposed mechanism is also consistent with the fact that esterification of the β-lactam carboxylate severely decreases the catalytic effect of the metal ions.
Another model system results from the combination of zinc ions and tris(hydroxymethyl)aminomethane (Tris) buffers in the pH range of 7.5–10.0. Thus, it has been reported that the Zn2+–Tris system is a very effective, true catalyst for the hydrolysis of penicillins.8,9 In addition, we have found in previous studies10–12 that the Zn2+–Tris system is also capable of efficiently hydrolyzing other β-lactams, such as clavulanic acid, which is a typical mechanism-based inhibitor of active-site serine β-lactamases (clavulanic acid is also a fairly good substrate of the zinc-β-lactamase from B. fragilis4). Kinetic analyses carried out by Schwartz8 have revealed that the mechanism for the Zn2+–Tris-assisted hydrolysis of benzylpenicillin involves a ternary complex formed by benzylpenicillin, Zn2+ and Tris, which can be stabilised by the chelating effect of Tris. In this complex, a zinc-bound hydroxyl group of Tris can become a powerful nucleophile by donating its proton before attacking the β-lactam carbonyl group intramolecularly. Hence, in the sense that the rate-determining step is a unimolecular process, the reaction between Zn2+–Tris and β-lactams can be considered a simplified model for the conversion of the enzyme–β-lactam complexes at the active site of the zinc-β-lactamases.
In this study, we have explored the influence of the β-lactam structure on the rate of the Zn2+-assisted hydrolysis in the presence of Tris. First, we confirmed that the hydrolysis of clavulanic acid obeys the general kinetic scheme involving a ternary complex. Then we addressed experimentally the kinetic influence of the β-lactam structure by determining the kobs values for a series of representative antibiotics including penicillins, cephalosporins, carbapenems and monobactams. The experimental results were complemented by carrying out quantum chemical calculations on cluster models representing the most likely complexes formed between Zn2+, the β-lactam models and the nucleophile. On the basis of these calculations, we discuss the modes of binding between the β-lactam nucleus and the metal ion, the possible pathways for the nucleophilic attack of the zinc-bound nucleophile on the β-lactam carbonyl, together with the kinetic influence of the β-lactam structure. Overall, the experimental and theoretical results complement each other well and give insight into the molecular details of the Zn2+-assisted hydrolysis process of β-lactams. To shed some light on the actual mode of binding between aztreonam and zinc-β-lactamases, we also carried out docking calculations. Finally, the relevance of all the results for zinc-β-lactamases is briefly discussed.
Methods
Materials
Aztreonam was obtained from Squibb & Sons, Inc. (Plainsboro, NJ, USA); Imipenem from Merck Sharp and Dohme (Rahway, NJ, USA); SCH 29482 from Schering Corporation (Blomfield, NJ, USA); Nocardicin A from Fujisawa Pharmaceutical Co. (Osaka, Japan); Moxalactam from Eli Lilly and Co. (Indianapolis, IN, USA) and Clavulanic acid, Amoxicillin, Cephaloglycin, Cephaloridine and Cephalothin from Antibióticos S. A. (León, Spain). These antibiotics were used as received. The structure of these antibiotics is shown in Scheme 1. Tris(hydroxymethyl)aminomethane (Tris) was a very pure grade from Sigma Chemical Co. Solutions of ZnCl2 were prepared as previously described.11 Other chemicals were commercial products of analytical grade.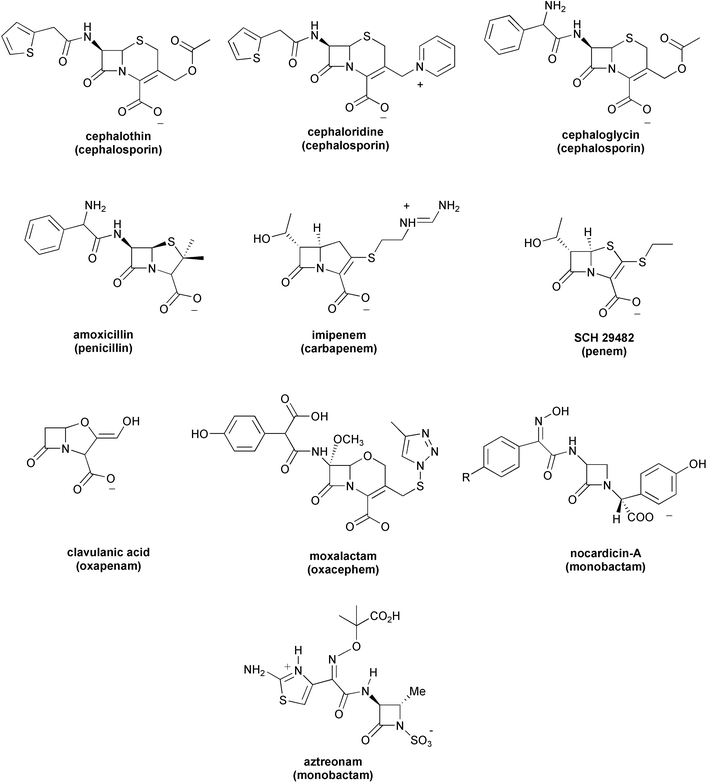 | ||
| Scheme 1 | ||
All the water used was purified by a Milli-Q-Reagent Water System (Millipore, Bedford, MA, USA). The solutions were freshly prepared and the pH measured at 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C using a WTW pHmeter (pH 526) equipped with a combined electrode with an integrated temperature sensor (Sentix 97T). All reactions were conducted at 35.0±0.1°C and the ionic strength was adjusted to 0.5 mol l−1 with sodium perchlorate.
°C using a WTW pHmeter (pH 526) equipped with a combined electrode with an integrated temperature sensor (Sentix 97T). All reactions were conducted at 35.0±0.1°C and the ionic strength was adjusted to 0.5 mol l−1 with sodium perchlorate.
Analytical procedure
The residual antibiotic concentration was determined using a reverse-phase high-performance liquid chromatographic (RP-HPLC) method. An Alliance® HPLC System liquid chromatograph from Waters (Mildford, MA, USA) equipped with a 2695 Separations Module and a 996 Photodiode Array Detector were used. The separation was carried out using a Phenomenex C18 (2) Luna column (5 µm; 150×4.60 mm). The analyses were carried out at 25 °C on a 20 µL injected volume. The mobile-phase characteristics and other chromatographic parameters are given in Table 1. The mobile phases were prepared fresh on the day of analysis and were filtered through a Millipore filter (0.45 µm pore size).
| Parameter | Antibiotic | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AZ | IM | SCH | NA | MX | CL | AM | CG | CR | CT | |
| Flow-rate/mL min−1 | 1.5 | 1.5 | 1 | 1 | 1.5 | 1 | 1.5 | 1.5 | 1 | 1.5 |
| Mobile phase | ||||||||||
| % 0.1 M sodium phosphate | 93 | 93 | 86.5 | 93 | 93 | 95 | 88 | 86.5 | 86.5 | 86.5 |
| % Methanol | 7 | 7 | 6.5 | 7 | 7 | 5 | 12 | 6.5 | 6.5 | 6.5 |
| % Isopropanol | 0 | 0 | 7 | 0 | 0 | 0 | 0 | 7 | 7 | 7 |
| λ/nm | 230 | 310 | 310 | 280 | 230 | 230 | 250 | 250 | 250 | 250 |
Kinetic procedure
Weighed amounts of β-lactam antibiotics were dissolved in the Tris buffer solutions to give concentrations of 1−5×10−4 mol dm−3. At appropriate time intervals aliquots of the solutions were each sealed in a glass vial and the reaction was blocked with ethylenediaminetetraacetic acid (EDTA; final concentration 2×10−4 mol dm−3), before analysis of the remaining β-lactam antibiotics.
In some cases where buffer capacity was too low, the pH of the kinetic solution during the reaction was maintained with a pH-stat (titrimeter assembly consisting of an E-614 Impulsomat, an E-655 Dosimat and an E-632 pHmeter from Metrohm, Herisau, Switzerland).
In all solutions the total Tris concentration greatly exceeded the reacting substrate concentration to maintain pseudo-first-order kinetics.
Quantum chemical calculations
Calculations were carried out with the Gaussian 98 system of programs.13 Stable structures were fully optimised and transition structures located at the B3LYP/6-31G* level.14–16 All the critical points were further characterised by analytic computation of harmonic frequencies at the same theoretical level. To further confirm the reaction paths on the potential energy surface connecting specific complexes and intermediates, intrinsic reaction coordinate (IRC) calculations followed by energy minimisation were also carried out at the B3LYP/6-31G* level (see below). Thermodynamic data (298 K, 1 bar) were computed using the B3LYP/6-31G* frequencies within the ideal gas, rigid rotor, and harmonic oscillator approximations. ΔGgas phase energies were obtained for all of the optimised species by combining the B3LYP/6-31G* electronic energies with the zero-point vibrational energy (ZPVE) values and thermal corrections.To take into account condensed phase effects on the kinetics and thermodynamics of the model systems, we used the united atom Hartree–Fock (UAHF) parameterisation17 of the polarisable continuum model (PCM),18 including both electrostatic and non-electrostatic solute–solvent interactions and simulating water as the solvent. The Gibbs solvation energies ΔGsolvation of all the critical structures were then computed from single-point B3LYP/6-31G* PCM-UAHF calculations on the B3LYP/6-31G* gas phase geometries. Addition of ΔGgas phase to the corresponding relative Gibbs solvation energies, ΔΔGsolvation, evaluated neglecting the change in the relative value of the thermal corrections when going from a vacuum to the solution, gives ΔGsolution for the structures studied.
Docking calculations
We employed the algorithm developed for AutoDock,19,20 which uses a Monte Carlo simulated annealing technique for the configurational exploration of enzyme–ligand complexes with a rapid energy evaluation using grid-based molecular interaction potentials built from van der Waals and electrostatic contributions. Aztreonam was docked at the static binding site of the B. cereus zinc-β-lactamase. During the docking process, the internal bonds of the aztreonam sidechains were allowed to rotate.The Jaguar program21 was used to optimise the molecular structure of aztreonam at the HF/6-31G* SCRF level.22 Then we obtained the electrostatically derived atomic charges23 for aztreonam, which were required for the docking calculations. The coordinates of the protein atoms were taken from the 1.85 Å crystal structure of B. cereus described by Carfi et al.24 (PDB ID code 1BME). This structure shows one fully and one partially occupied zinc site, and for our purposes we deleted the second zinc ion and a carbonate anion from the coordinate file in order to model the native form of the mononuclear B. cereus enzyme. All the crystallographic water molecules were also removed from the PDB file except the Wat1 molecule near the Zn1 ion. Hydrogen atoms were added to the protein system using the LEaP program,25 while atomic charges were taken from the AMBER force field.26
Results
Kinetics of the reaction between clavulanic acid and Zn2+–tris
As shown in previous work, the reaction of clavulanic acid with excess Tris and in the absence of metal ions follows pseudo-first-order kinetics with respect to the concentration of Tris (denoted [T]).12 In the presence of both Tris buffer (up to 0.20 M) and Zn2+ ions at concentrations ranging up to 5×10−5 M, clavulanic acid was also observed to degrade according to pseudo-first-order kinetics, where the rate constant kobs for the latter process is a linear function of the metal ion concentration. For this study, we reinvestigated the kinetics of the Zn2+–Tris system reacting with lithium clavulanate at different pH. Thus, Fig. 1 shows the dependence of kobs on the total concentration of Tris at pH 7.50, 8.00, 8.50, 9.00 and 9.50 for the decomposition of the β-lactam in solutions containing the same concentration of Zn2+. The catalytic action of the free Tris species (i.e., not coordinated to the Zn2+ ions) can be subtracted from kobs by means of eqn. (1): | kz=kobs−kTris[T] | (1) |
=0.114 M−1 min−1 assuming a pKa value for Tris of 8.01; see ref. 8). Fig. 1 also plots the values calculated for the rate constant kz.
 | ||
| Fig. 1 Dependence of the constants kobs
(○) and kz
(□) on the concentration of Tris at different pHs for the decomposition of lithium clavulanate (5.0×10−4 M) in the presence of Zn2+ 5.0×10−6 M, 0.5 M ionic strength (NaClO4) at a temperature of 308±0.1 K. | ||
The curves represented in Fig. 1 show that kobs and kz exhibit close-lying maxima at a total concentration of Tris between 0.015 and 0.050 M. At high pH, when the concentration of Tris is increased (>0.050 M) the Tris-only route (i.e., kTris) becomes more evident. However, the rate for β-lactam decomposition tends to decrease at high [T], most probably because the Zn2+ ions are sequestered by the excess of Tris molecules, which inhibits the catalytic effect of the metal. Overall, the data in Fig. 1 show that the combination of Zn2+ ion and Tris buffers notably increases the rate of β-lactam degradation. It is interesting to note that most of the observed degradation of the clavulanate anion near physiological pH (7.50) proceeds through reaction with the Zn2+–Tris complex (see Fig. 1). On the other hand, the clavulanate anion reacts faster at higher pH given that, under alkaline conditions, most of the Tris molecules (pKa=8.01) would be in their neutral form and, therefore, clavulanate could be attacked more readily by either the Tris hydroxyl or the free amino groups through direct or Zn2+-assisted mechanisms.
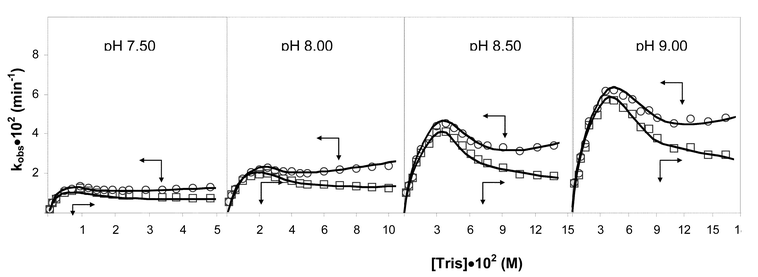
All of the points for each pH were utilised to determine the best fit to eqn. (2), which was derived from a mechanism proposed by Schwartz.8
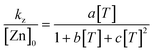 | (2) |
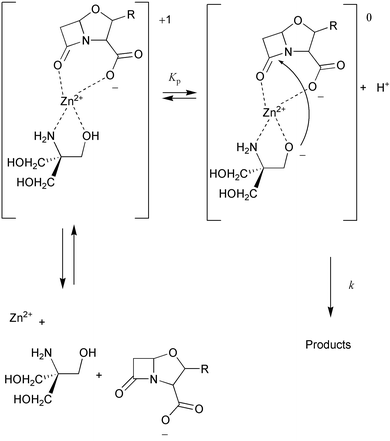
Following the mechanism proposed by Schwartz, the rate of reaction would be proportional to the concentration of a ternary Zn2+–Tris–clavulanate (ZnTC°) complex:
| Rate=kz[C]=k[ZnTC°] | (3) |
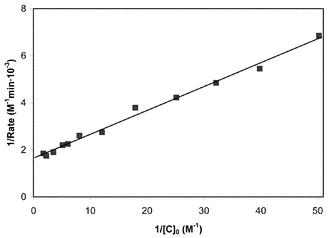 | ||
| Fig. 2 Double-reciprocal plot of inverse rate vs. inverse clavulanate ion concentration at pH 8.00 and for initial Zn2+ and Tris concentrations of 1.0×10−6 M and 0.050 M, respectively. | ||
 | ||
| Scheme 2 | ||
For benzylpenicillin, complete kinetic analyses and rate constant derivations based on the mechanism displayed in Scheme 2 have been previously reported by Schwartz.8 We carried out an analogous quantitative treatment of the kinetic data for the reaction of clavulanic acid, which is fully detailed in the Electronic supplementary information (ESI). These analyses indicate that the formation of the ternary complex must occur when a binary Zn2+–Tris complex binds to one clavulanate anion. The formation constants for the Zn2+–Tris chelates are K1=182 for Zn2+–Tris and K2=25 for Zn2+–Tris2.8 Subsequently, the resultant ternary complex loses a proton, which is most probably released from a zinc-bound hydroxyl group of Tris. The zinc-bound nucleophile attacks the β-lactam carbonyl group, leading to rupture of the β-lactam ring. The expected product would be a Tris ester.8 These esters are known to hydrolyse rather readily and are also subject to aminolysis.27–29 The values obtained for the constants at work in the mechanism proposed by Schwartz for the degradation of benzylpenicillin in the presence of Zn2+–Tris are very similar to our values for the degradation of clavulanic acid under the same conditions. According to our results, the estimated value of the kinetic constant k for the acylation step is quite high, 4×103 min−1, which explains the rapid decomposition of the clavulanate anion in the presence of Zn2+–Tris (k=2.5×104 min−1 for benzylpenicillin8). The fact that both benzylpenicillin and clavulanic acid obey the same kinetic scheme despite their structural differences suggests that the molecular mechanism of β-lactam alcoholysis catalysed by the Zn2+–Tris system may be common to the various families of β-lactam antibiotics.
Influence of the β-lactam structure on the reaction with Zn2+-tris
As mentioned in the Introduction, the β-lactam carboxylate group must interact directly with the metal ion, since its esterification produces β-lactams, which are not susceptible to metal-catalysed hydrolysis.27 Of course, the Zn2+⋯OOC-β-lactam interaction can be influenced by the geometrical orientation of the β-lactam carboxylate group (N-sulfonic group in monobactams) with respect to the scissile amide bond, which varies along the spectrum of β-lactam antibiotics.To determine the kinetic influence of the β-lactam structure on their reactivity with the Zn2+–Tris system, we obtained the kobs values at different metal concentrations for a series of β-lactam compounds including imipenem, SCH29482, amoxicillin, clavulanic acid, moxalactam, cephaloglycin, aztreonam, and nocardicin A (see Scheme 1). The results are summarised in Table 2 and Fig. 3.
![Effect of concentration of Zn2+ on the observed rate constant for the decomposition of β-lactam antibiotics. Experimental conditions: pH = 8.00, [Tris] = 0.050 M, [β-lactam] = 1 × 10−3 M, 0.5 M ionic strength (NaClO4), temperature = 308 ± 0.1 K. (a) Decomposition of imipenem (□), amoxicillin (+), SCH29482 (○), clavulanic acid (*), cephaloglycin (⊡), moxalactam (●), aztreonam (▲) and nocardicin A (△). (b) Decomposition of cephaloglycin (×), cephaloridine (●) and cephalothin (■).](/image/article/2004/NJ/b306799h/b306799h-f3.gif) | ||
| Fig. 3 Effect of concentration of Zn2+ on the observed rate constant for the decomposition of β-lactam antibiotics. Experimental conditions: pH=8.00, [Tris]=0.050 M, [β-lactam]=1×10−3 M, 0.5 M ionic strength (NaClO4), temperature=308±0.1 K. (a) Decomposition of imipenem (□), amoxicillin (+), SCH29482 (○), clavulanic acid (*), cephaloglycin (⊡), moxalactam (●), aztreonam (▲) and nocardicin A (△). (b) Decomposition of cephaloglycin (×), cephaloridine (●) and cephalothin (■). | ||
We see in Fig. 3 that β-lactam antibiotics that possess a five-membered ring fused with the β-lactam ring are clearly more reactive than other antibiotics such as cephalosporins and monobactams. This is not entirely unexpected since cephalosporins and monobactams are generally thought to be less reactive because they present a less strained β-lactam amide bond. We note, however, that the intrinsic reactivity of aztreonam towards alkaline hydrolysis is similar (within a factor of two) to that of benzylpenicillin and carbapenems.30 Hence, other electronic and structural factors related to the interaction between Zn2+–Tris and the β-lactam could well explain the results shown in Fig. 3. In fact, the bicyclic antibiotics, which have carboxylate groups with a similar orientation relative to the amide bond, may coordinate the metal ion more efficiently than monobactams throughout the reaction.
For β-lactam antibiotics within the same family, the number and kind of side chains attached to the β-lactam nucleus influences their relative activity. For example, cephaloridine, which has a positively charged leaving group at the C3 position, reacts faster than an analogous cephalosporin (cephalothin) with a neutral group [see data plotted in Fig. 3(b)]. No differences in reactivity are observed, however, between cephalothin and cephaloglycin, which differ only in the side chain at position 7.
Model systems used in the quantum chemical calculations
To complement the kinetic analyses and experimental data on the Zn2+–Tris system, we employed density functional theory (DFT) methodologies to obtain molecular structures and energies for a series of model systems. More specifically, the DFT calculations were carried out in order to (a) characterise the reactive modes of binding between the β-lactam nucleus and the Zn2+ ion, (b) find out the possible pathways for the hydrolysis process, and (c) explain the large kinetic effect exerted by the β-lactam structure. To this end, we studied the structure and reactivity of the cluster models IMI and MONO as shown in Scheme 3.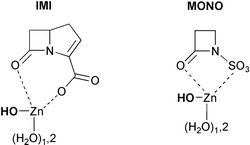 | ||
| Scheme 3 | ||
In the model system IMI, the β-lactam moiety represents the bicyclic structure of the most reactive antibiotics in the presence of Zn2+–Tris (i.e., carbapenems) while the model system MONO is related to the antibiotic aztreonam, which is less reactive than carbapenems by a factor of ∼103 (see Table 2). Both models assume that the Zn2+ ion coordinates simultaneously to some water molecules and to the β-lactam carboxylate or N-sulfonate group. We note that, although Tris molecules were not considered to keep the theoretical models within tractable limits, the cluster models IMI and MONO correspond formally to ternary complexes involving the β-lactam, the metal ion and the zinc-bound nucleophile, according to the experiments. One of the zinc-bound water molecules is already deprotonated in the cluster models, given that we concentrate on understanding the molecular mechanism of the hydrolysis reaction (the last mechanistic step in Scheme 2).
All the structures (minima and transition structures) that were located on the B3LYP/6-31G* potential energy surface are shown in Figs. 4–8. The corresponding free energy profiles in solution (ΔGsolution) are also represented in Figs. 5–8.
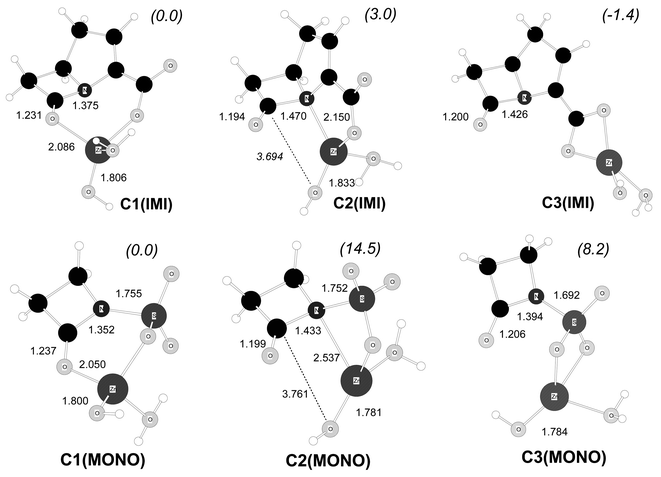 | ||
| Fig. 4 B3LYP/6-31G* optimised structures for some Zn2+–β-lactam model complexes. Distances in Å. Relative free energies in solution (kcal mol−1) are indicated in parentheses. | ||
In the structure C1(IMI), the Zn2+ion bridges the β-lactam carbonyl and carboxylate groups. In C2(IMI) the metal ion is coordinated to both the endocyclic N atom and the carboxylate group. In addition, we also studied the complex C3(IMI) in which the β-lactam carboxylate binds to the Zn2+ ion in a bidentate manner. For the MONO system, analogous modes of binding involving the N-sulfonate group were also characterised by locating the corresponding complexes C1(MONO), C2(MONO) and C3(MONO) (See Fig. 4).
The modes of β-lactam–Zn2+ binding represented in Fig. 4 have been postulated in previous work in order to explain the catalysis exerted by metal ions.6 However, our calculations give new insight about the structure and relative stability of these complexes. For example, the carbapenem-like complexes, C1(IMI), C2(IMI) and C3(IMI), are quite close in free energy. The bidentate adduct C3(IMI) is predicted to be the most stable one, being only 1.4 and 4.4 kcal mol−1 below C1(IMI) and C2(IMI), respectively. Taking into account the highly labile kinetic nature and the low thermodynamic stability of the Zn2+ complexes,31 we expect that the ΔG energy barriers for the C1(IMI)↔C2(IMI)↔C3(IMI) interconversion processes in solution would not be great and, therefore, these complexes would interconvert rapidly. In contrast, a more abrupt free energy profile for the binding of monobactams to Zn2+ ions is obtained from the C1(MONO), C2(MONO) and C3(MONO) structures: the C1(MONO) complex is 14.5 and 8.2 kcal mol−1 more stable than C2(MONO) and C3(MONO), respectively. Thus, the mode of binding in which the metal is simultaneously bound to the β-lactam carbonyl and N-sulfonate groups would predominate in solution.
The relative ΔG values in Fig. 4 show that the structure of the β-lactam determines the stability of its complexes with Zn2+. On the one hand, the bicyclic skeleton of carbapenems seems well suited for binding the Zn2+ ions through different coordination modes. On the other hand, the N-sulfonate group imposes more stringent geometrical and electronic restrictions in the formation of the β-lactam–Zn2+ complexes. Thus, C2(MONO) presents a strained mode of coordination through a four-membered ring in which the Zn⋯N(β-lactam) distance is quite long (2.537 Å). In C3(MONO), binding through the N-sulfonate group perturbs the N–SO3− functionality since the N–S distance is around 0.06 Å shorter than in the other complexes.32
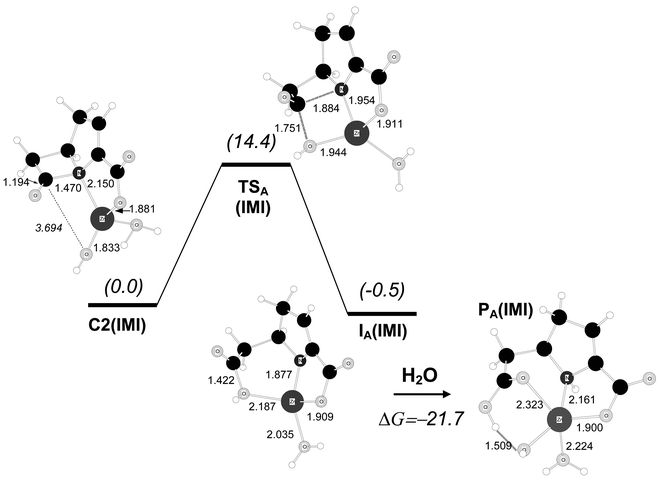 | ||
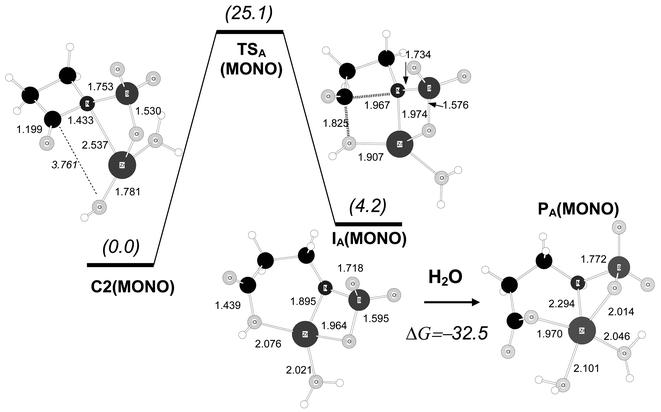
| Fig. 5 B3LYP/6-31G* optimised structures for the hydrolysis reaction of the carbapenem model compound starting from complex C2(IMI). Distances in Å. Relative free energies in solution (kcal mol−1) are shown in parentheses. | ||
 | ||
| Fig. 6 B3LYP/6-31G* optimised structures for the hydrolysis reaction of the monobactam model compound starting from complex C2(MONO). Distances in Å. Relative free energies in solution (kcal mol−1) are shown in parentheses. | ||
From C2(IMI), the formation of the C–O bond and cleavage of the β-lactam ring takes place in a single mechanistic step passing through the transition structure TSA(IMI), which has C–O/C–N distances for the forming/breaking bonds of 1.751/1.884 Å. In terms of the B3LYP/6-31G* free energies in solution, TSA(IMI) is 14.4 kcal mol−1 above C2(IMI) and 18.8 kcal mol−1 above the most stable complex C3(IMI). TSA(IMI) is connected with an intermediate species, IA(IMI), in which the β-lactam ring is completely cleaved while the carboxylic group and the negatively charged N atom are bound to the Zn2+ ion. This intermediate has a similar stability to that of the initial complex C2(IMI) with a relative ΔG value of −0.5 kcal mol−1.
Completion of the hydrolysis reaction requires that the β-lactam N atom accepts a proton, most probably from a water molecule. Several pathways, not detailed here, are possible for this process (e.g., the proton may come from bulk water or a zinc-coordinated water molecule). To estimate the thermodynamics of the protonation step, we optimised a product complex PA(IMI)
(see Fig. 5) that was obtained from IA(IMI) by adding an extra water molecule. In the course of the energy minimisation, the extra water molecule became coordinated to the zinc ion and donated one proton to the N atom. The ΔG value of PA(IMI) with respect to IA(IMI)+H2O is −21.7 kcal mol−1, thus showing that protonation of the IA(IMI) intermediate is very favourable thermodynamically.
For the monobactam system, a hydrolysis mechanism analogous to that of the IMI system was also studied (see Fig. 6). Most interestingly, the calculated transition structure and intermediate [TSA(MONO) and IA(MONO) in Fig. 6] are much less stable than those for the carbapenem model, resulting in a ΔG barrier for the C2(MONO)→TSA(MONO) process of 25.1 kcal mol−1. In contrast, the overall reaction process leading to PA(MONO) is favourable [the ΔG value of PA(MONO) with respect to IA(MONO)+H2O is −32.5 kcal mol−1].
By taking into account the ΔG value in going from C1(MONO) to C2(MONO), the global ΔG barrier for the hydrolysis of the monobactam model amounts to 39.6 kcal mol−1. This high energy barrier can be ascribed to a less favourable (i.e., more strained) β-lactam–Zn2+ binding along the reaction coordinate even though location of the negative charge on the leaving N atom results in short Zn–N contacts of ∼1.9 Å at TSA(MONO) and IA(MONO). However, the corresponding N–Zn–O bond angles are very acute: 75.0° and 79.0°, respectively. In contrast, the same angles in the carbapenem structures TSA(IMI) and IA(IMI) are close to 90 degrees, 89.3° and 94.6°, respectively.
The calculations summarised in Figs. 5–6 confirm that the nature of the β-lactam has a large kinetic impact on the Zn2+-assisted hydrolysis processes when starting with complexes such as C2(IMI) and C2(MONO). In fact, the ΔG barriers computed suggest that carbapenems would degrade rapidly whereas monobactams would hydrolyse very slowly. However, it must be noted that other reaction mechanisms starting from different pre-reactive complexes and/or involving the participation of ancillary water molecules could be operative. To further investigate this point, we studied a second mechanistic route starting at pre-reactive complexes in which the zinc ion bridges the carbonyl group and the carboxylate/N-sulfonate groups of the carbapenem/monobactam models. This mode of binding is quite favourable for both β-lactam structures (see Fig. 4). In the calculations for this second mechanism, two more water molecules were included with respect to the tetrahedral complexes C1(IMI) and C1(MONO). After geometry optimisation, one of these extra waters coordinates directly to the Zn2+ ion, which now has fivefold coordination, while the other one is situated in the second coordination sphere of the metal in an appropriate position to assist the proton transfer to the leaving N atom (see CB(IMI) and CB(MONO) in Figs 7–8).
Fig. 7 represents the transition structures and minima involved in the hydrolysis of the initial carbapenem complex CB(IMI). This mechanism proceeds in a stepwise manner, passing through a tetrahedral intermediate. According to IRC and energy minimisation calculations on the B3LYP/6-31G* potential energy surface, complex CB(IMI)
(with fivefold coordination around the Zn2+ ion) is connected with the transition structure TS1B(IMI)
(with tetrahedral zinc coordination). At TS1B(IMI) the nucleophile is a water molecule that was initially bound to the Zn2+ ion in the pre-reactive complex. The transition vector of TS1B(IMI) is dominated by the formation of the C–O bond (1.725 Å) while the attacking water molecule is hydrogen-bonded to the zinc-coordinated hydroxide anion and the ancillary water molecule (see Fig. 7). From TS1B(IMI) the tetrahedral intermediate IB(IMI) is reached after having completed the formation of the C–O bond with simultaneous proton transfer from the nucleophilic water molecule to the zinc-bound hydroxide. At IB(IMI) the Zn2+ ion shows tetrahedral coordination, the amide bond C–N is quite elongated (C–N=1.580 Å) and the hydroxyl group is linked to the endocyclic N atom via a one-water bridge. The following transition structure, TS2B(IMI) in Fig. 7, corresponds to β-lactam ring rupture with simultaneous proton transfer from the hydroxyl group to the leaving amino group assisted by the ancillary water molecule. TS2B(IMI) leads to the product complex PB(IMI) in which the Zn2+ ion is coordinated by two carboxylate groups and the amino group of the degraded carbapenem model. The free energy profile for this mechanism is quite low: the ΔG values in kcal mol−1 are 0.0 [CB(IMI)]→10.7 [TS1B(IMI)]→3.3 [IB(IMI)]→12.7 [TS2B(IMI)]→−38.4 [PB(IMI)]. Hence, the rate-determining step is the cleavage of the tetrahedral intermediate via TS2B(IMI). Though not directly comparable, we note that the ΔG barrier (12.7 kcal mol−1) is ∼6 kcal mol−1 lower than that for the mechanism involving direct nucleophilic attack from the zinc-bound hydroxide at C2(IMI).
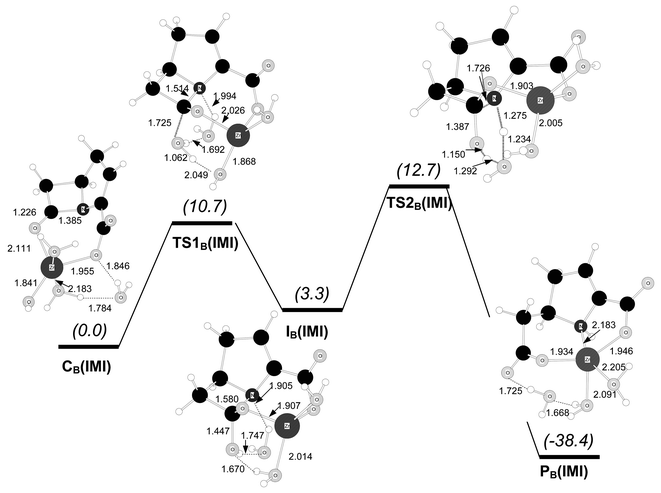 | ||
| Fig. 7 B3LYP/6-31G* optimised structures for the hydrolysis reaction of the carbapenem model compound starting from complex CB(IMI). Distances in Å. Relative free energies in solution (kcal mol−1) are shown in parentheses. | ||
For the monobactam model, the initial complex CB(MONO) shows a mode of binding between Zn2+ and the β-lactam analogous to that found in CB(IMI). For example, the Zn2+ ion is nearly coplanar to the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N amide group both in CB(MONO) and CB(IMI)
(the N–SO3− group is also coplanar to the OC–N group). The hydrolysis of CB(MONO) with proton transfer assisted by the ancillary water molecule takes place in a concerted fashion. At TSB(MONO) the nucleophilic attack is quite advanced (C–O=1.467 Å), the endocylic C–N bond is clearly elongated (C–N=1.674 Å) and the two protons involved in the assisted proton transfer to the β-lactam N atom are “in flight”
(see Fig. 8). The computed ΔG barrier is 25.0 kcal mol−1. On the one hand, this ΔG value indicates that hydrolysis of monobactams from complex CB(MONO) is clearly less favoured by ∼12 kcal mol−1 than the equivalent pathway for carbapenems. On the other hand, on comparison with the reaction mechanism from the tetrahedral complexes C1(MONO) and C2(MONO), it turns out that the hydrolysis of the monobactams starting at complexes like CB(MONO) would be more favourable. This is well understood in terms of the coordination geometry around the Zn2+ ion, which is not strained in the latter pathway in contrast with the C1(MONO)→C2(MONO)→TSA(MONO) route (see above).
C–N amide group both in CB(MONO) and CB(IMI)
(the N–SO3− group is also coplanar to the OC–N group). The hydrolysis of CB(MONO) with proton transfer assisted by the ancillary water molecule takes place in a concerted fashion. At TSB(MONO) the nucleophilic attack is quite advanced (C–O=1.467 Å), the endocylic C–N bond is clearly elongated (C–N=1.674 Å) and the two protons involved in the assisted proton transfer to the β-lactam N atom are “in flight”
(see Fig. 8). The computed ΔG barrier is 25.0 kcal mol−1. On the one hand, this ΔG value indicates that hydrolysis of monobactams from complex CB(MONO) is clearly less favoured by ∼12 kcal mol−1 than the equivalent pathway for carbapenems. On the other hand, on comparison with the reaction mechanism from the tetrahedral complexes C1(MONO) and C2(MONO), it turns out that the hydrolysis of the monobactams starting at complexes like CB(MONO) would be more favourable. This is well understood in terms of the coordination geometry around the Zn2+ ion, which is not strained in the latter pathway in contrast with the C1(MONO)→C2(MONO)→TSA(MONO) route (see above).
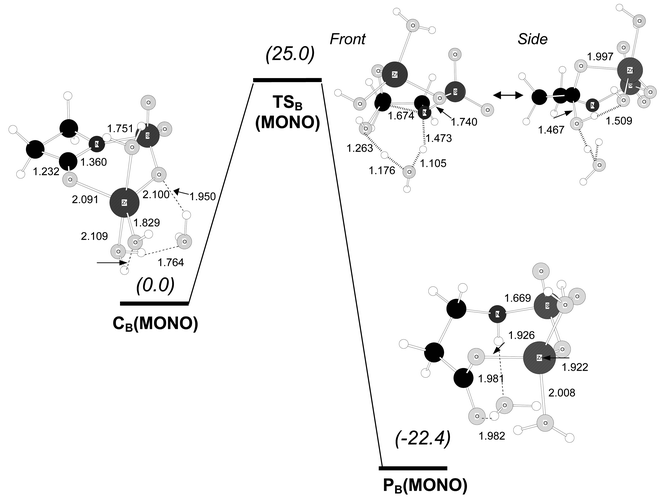 | ||
| Fig. 8 B3LYP/6-31G* optimised structures for the hydrolysis reaction of the carbapenem model compound starting from complex CB(MONO). Distances in Å. Relative free energies in solution (kcal mol−1) are shown in parentheses. | ||
Discussion
The experimental kinetic results presented in this work confirm that the Zn2+–Tris system has an important catalytic effect on the degradation of many β-lactam antibiotics including carbapenems, oxapenams, oxacephems, penicillins and cephalosporins. For clavulanic acid, the detailed kinetic analyses indicate that the main mechanistic route for the Zn2+–Tris-assisted degradation implies the formation of “ternary” complexes that bring together the Zn2+ ion, the β-lactam and the deprotonated nucleophile prior to nucleophilic attack. This result is in agreement with the original work on benzylpenicillin hydrolysis.8,9The fast catalysis and the kinetic role of the pre-reactive complexes make the Zn2+–Tris system an appropriate model system for evaluating the inherent reactivity of different β-lactam antibiotics with Zn2+ complexes in aqueous solution. In effect, we found that the relative activity of a large set of antibiotics spans a broad range of 4 orders of magnitude: carbapenems, penicillins, and oxapenams are highly reactive in the presence of Zn2+–Tris, cephalosporins exhibit an intermediate reactivity and monobactams are clearly less reactive.
As mentioned in the Introduction, most metallo-β-lactamases are capable of hydrolysing most β-lactams except monobactams. Interestingly, our experimental model system partially mimics the substrate spectrum of the metallo-β-lactamases given that the Zn2+–Tris system also exhibits poor hydrolysis of monobactams. However, to find out if the structural effects controlling the rate of the reaction of monobactams with Zn2+–Tris are also relevant to the metallo-β-lactamases, a molecular description of the model systems is required. Here, the quantum chemical calculations complement the experimental observations well.
The theoretical work was carried out on cluster models that take into account only the nucleus of carbapenems and monobactams bearing the N–SO3− group. To further simplify the theoretical approach, water molecules (and one hydroxide anion), instead of Tris, were considered. By computational examination of these models, we concentrated on the structural and energetic influences of the β-lactam structure in their reaction with the Zn2+ ions in aqueous solution. Nevertheless, we note that the mode of binding between the β-lactam and the Zn2+ ions in our water-containing models can also occur if one Tris molecule exchanges with two water molecules at the Zn2+ position.
The DFT calculations indicate that both the thermodynamic preferences for the mode of binding between the β-lactam and Zn2+ and the kinetic energy profile for the hydrolysis of the β-lactam are largely affected by the structure of the β-lactam. For example, we observed that the donor functionalities in carbapenem-like structures (carbonyl, the endocyclic N atom and the exocyclic carboxylate) are spatially adapted to ligate the Zn2+ cation in three coordination modes with a similar stability. Therefore, we propose that, during the reaction of carbapenem-like β-lactams with Zn2+–Tris or similar systems, different “ternary complexes” could be in rapid interconversion. Moreover, we found that at least two catalytic routes for the hydrolysis of the β-lactam carbepenem model starting from different complexes are energetically viable with ΔG barriers of 13–19 kcal mol−1. In the first one, the Zn2+ ion binds simultaneously to the endocyclic N atom and the carboxylate group while a zinc-bound hydroxyde group (methoxide in Zn2+–Tris) attacks the carbonylic C atom. In the second route, the carbonyl and carboxyl groups of the β-lactam are bridged through the metal ion along the reaction coordinate and the nucleophile is a water molecule (hydroxymethyl in Zn2+–Tris) that was initially ligated to the Zn2+ ion. In contrast to the carbapenem model, the N–SO3− and carbonyl groups of the monobactam compound chelate the Zn2+ ion quite “rigidly” in comparison with the other modes of coordination. Moreover, the N–SO3− group creates ring strain at the key transition structures for hydrolysis. As a consequence, only one mechanism for the Zn2+-assisted hydrolysis is possible with a relatively high ΔG barrier (∼25 kcal mol−1). These theoretical results rationalise satisfactorily the experimental fact that aztreonam is much less reactive than carbapenems in the presence of the Zn2+–Tris system.
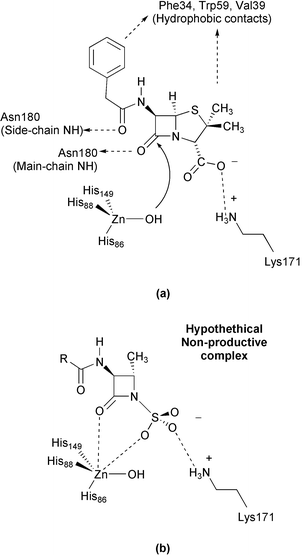
Can our results on the hydrolysis reaction of model systems give some insight into the interaction of monobactams with metallo-β-lactamases? Before attempting to answer this question, it must be noted that metallo-β-lactamases have accessible active sites that can accommodate β-lactam substrates with very different side chains attached to the β-lactam nucleus.2 Based on the results of docking and molecular dynamics simulations of the mononuclear metallo-β-lactamase from Bacillus cereus complexed with benzylpenicillin (see Scheme 4a),33 it has been suggested that substrate specificity stems from H-bonds and salt bridge interactions of the β-lactam with major conserved residues, whereas the Zn complex at the active site merely provides a desolvated and properly oriented “hard” nucleophile. Intriguingly, the B. cereus enzyme can also accommodate aztreonam molecules at its active site, according to our docking analyses (see Fig. 9). In the docked model, the monobactam nucleus is well orientated for catalysis: the β-lactam carbonyl points towards the Zn site [Zn⋯OC(monobactam)=3.4 Å; Zn⋯O3S(monobactam)=5.0 a] while the N-sulfonate gives a salt bridge with Lys171 (Nζ@Lys171⋯O3S∼3.0 Å). Thus, it may be reasonably expected that the lack of monobactam activity in the metallo-β-lactamases could be related to the particular structure of the monocyclic nucleus. Although protein flexibility and solvent dynamics must be considered to fully characterise the aztreonam-binding determinants, we note that the “three-pronged” N–SO3− group might interact simultaneously with the nearby lysine and the Zn ion [see Scheme 4(b)]. In this hypothetical configuration, the monobactam coordination around the zinc centre could be too “rigid” to give enzymatically productive aztreonam–enzyme complexes.
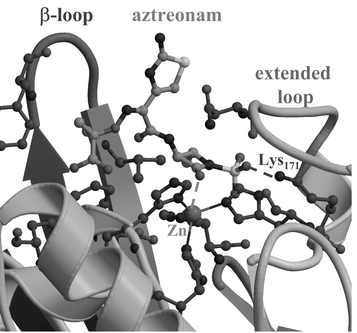 | ||
| Fig. 9 Docking model of a complex between aztreonam and the B. cereus enzyme. | ||
 | ||
| Scheme 4 | ||
Acknowledgements
The authors are grateful to the CICyT (Spain) for a generous allocation of computer time at the CESCA and the CIEMAT. Financial support by the Spanish Ministry of Science and Technology (MCyT) via grant SAF2001-3526 is also acknowledged.References
- D. M. Livermore and N. Woodford, Curr. Opin. Microbiol., 2000, 3, 489 CrossRef CAS.
- Z. Wang, W. Fast, A. M. Valentine and S. J. Benkovic, Curr. Opin. Chem. Biol., 1999, 3, 614 CrossRef CAS.
- K. Bush, Clin. Infect. Dis., 1998, S48 CAS.
- C. Prosperi-Meys, G. Llabres, D. de Seny, R. Paul-Soto, M. Hernández-Valladares, N. Laraki, J. M. Frère and M. Galleni, FEBS Lett., 1999, 443, 109 CrossRef CAS.
- G. M. Rossolini, N. Franceschini, M. L. Riccio, P. S. Mercuri, M. Perilli, M. Galleni, J.-M. Frère and G. Amicosante, Biochem. J., 1998, 332, 145 CAS.
- M. I. Page, in The Chemistry of β-lactams, ed. M. I. Page, Blackie Academic and Professional, New York, 1992, pp. 129–147 Search PubMed.
- P. J. Montoya-Pelaez and R. S. Brown, Inorg. Chem., 2002, 41, 309 CrossRef CAS.
- M. A. Schwartz, Bioorg. Chem., 1982, 11, 4 CAS.
- H. Tomida and M. A. Schwartz, J. Pharm. Sci., 1983, 72, 331 CAS.
- J. Martín, R. Méndez and T. Alemany, J. Chem. Soc., Perkin Trans. 2, 1989, 223 RSC.
- J. Martín, R. Méndez and F. Salto, J. Chem. Soc., Perkin Trans. 2, 1989, 227 RSC.
- J. Martín, R. Méndez and F. Salto, J. Chem. Soc., Perkin Trans. 2, 1990, 43 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, P. Salvador, J. J. Dannenberg, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, GAUSSIAN 98 (Revision A.10), Gaussian, Inc., Pittsburgh, PA, 2001 Search PubMed.
- A. D. Becke, in Exchange-Correlation Approximation in Density-Functional Theory, ed. D. R. Yarkony, World Scientific, Singapore, 1995 Search PubMed.
- W. J. Hehre, L. Radom, P. v. R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, John Wiley & Sons, New York, 1986 Search PubMed.
- V. A. Rasolov, J. A. Pople, M. A. Patner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223 CrossRef CAS.
- V. Barone, M. Cossi and J. Tomasi, J. Chem. Phys., 1997, 107, 3210 CrossRef CAS.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027 CrossRef CAS.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639 CrossRef CAS.
- D. S. Goodsell and A. J. Olson, Proteins: Struct. Funct. Genet., 1990, 8, 195 CAS.
- JAGUAR, 4.1, Schrodinger Inc., Portland, OR, USA, 1998 Search PubMed.
- D. J. Tannor, B. Marten, R. Murphy, R. A. Friesner, D. Sitkoff, A. Nicholls, M. Ringnalda, W. A. Goddard III and B. Honig, J. Am. Chem. Soc., 1994, 116, 11875 CAS.
- B. H. Besler, K. M. Merz, Jr. and P. A. Kollman, J. Comput. Chem., 1990, 11, 431 CrossRef CAS.
- A. Carfi, E. Duée, M. Galleni, J. M. Frère and O. Dideberg, Acta. Crystallogr. Sect. D, 1998, 54, 313 CrossRef.
- C. Schafmeister, W. S. Ross and V. Romanovski, LEaP, University of California San Francisco (UCSF), 1995 Search PubMed.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, Jr., D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179 CrossRef CAS.
- H. Bungaard and C. Larsen, Arch. Pharm. Chem. Sci. Educ, 1978, 6, 184 Search PubMed.
- C. H. Schneider and A. L. de Weck, Immunochemistry, 1967, 4, 331 Search PubMed.
- D. E. Tutt and M. A. Schwartz, J. Am. Chem. Soc., 1971, 93, 767 CrossRef CAS.
- M. I. Page, Acc. Chem. Res., 1984, 17, 144 CrossRef CAS.
- H. Vahrenkamp, Acc. Chem. Res., 1999, 32, 589 CrossRef CAS.
- N. Díaz, D. Suárez and T. L. Sordo, Helv. Chim. Acta, 2002, 85, 206 CrossRef CAS.
- N. Díaz, D. Suárez and K. M. Merz, Jr., J. Am. Chem. Soc., 2001, 123, 9867 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: quantitative analysis of the kinetic data for the reaction of clavulanic acid. See http://www.rsc.org/suppdata/nj/b3/b306799h/ |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2004 |