An integrated approach to the study of the recognition of guests containing CH3 and CH2 acidic groups by differently rigidified cone p-tert-butylcalix[4]arene derivatives†
Giuseppe
Arena
*a,
Annalinda
Contino
a,
Elisa
Longo
a,
Giuseppe
Spoto
a,
Arturo
Arduini
b,
Andrea
Pochini
*b,
Andrea
Secchi
b,
Chiara
Massera
c and
Franco
Ugozzoli
*c
aDipartimento di Scienze Chimiche, Università di Catania, Viale Andrea Doria 6, I-95125, Catania, Italy. E-mail: garena@mbox.unict.it; Fax: +39 095 337678; Tel: +39 095 7385071
bDipartimento di Chimica Organica e Industriale, Università di Parma, Parco area delle Scienze 17/a, I-43100, Parma, Italy. E-mail: andrea.pochini@unipr.it; Fax: +39 0521 905472; Tel: +39 0521 905408
cDipartimento di Chimica Generale e Inorganica Chimica Analitica Chimica Fisica, Università di Parma, Parco Area delle Scienze 17/A, 43100, Parma, Italy. E-mail: ugoz@unipr.it; Fax: +39 0521 905556; Tel: +39 0521 905417
First published on 29th October 2003
Abstract
The efficiency and selectivity of calix[4]arenes, having different rigidities, in recognizing CH3X and CH2XY guests in apolar media have been investigated by 1H NMR spectroscopy. 1,3-Dipropoxy-p-tert-butylcalix[4]arene (1) turns out to be less selective than the comparatively more rigid biscrown-3-p-tert-butylcalix[4]arene (2). To obtain further information on these recognition processes, both calorimetric and structural studies have been performed in solution and in the solid state, respectively. The calorimetric study shows that the inclusion process is enthalpically driven and entropically unfavoured in all cases. The favourable enthalpic contribution mainly results from specific host–guest CH–π interactions. Methyl substituted guests show a less unfavourable entropic contribution. Solid state structural determinations of the complexes help in explaining these results.
Introduction
Molecular recognition phenomena play a key role in several biological processes where a natural host, thanks to the particular chemical and structural information stored in its structure, is able to selectively recognise a complementary substrate (guest). Several studies have been carried out over the last couple of decades to disclose the nature and role played by host–guest intermolecular weak forces that drive these processes. In this context, remarkable achievements have been obtained in several instances by studying the molecular recognition processes that take place between specifically designed artificial receptors and simple (charged or neutral) guests. Nevertheless, basic and systematic studies carried out using different and complementary techniques able to provide a deeper understanding of the relative magnitude of the different forces (factors) that drive these processes are still lacking, especially in those cases where the energy gain associated with complex formation is of few kJ per mole.Calixarenes,1 thanks to the arrangement of their aromatic rings, possess an electron rich cavity, that is suitable for the inclusion of neutral guests of complementary size. Although the complexation of neutral guests by water soluble calixarenes is well documented,2 a systematic study of the factors influencing the stability of calixarene complexes with neutral organic molecules in apolar media has been undertaken only recently. Arduini et al. have synthesized several calix[4]arenes, rigidified in a cone conformation by short ethereal bridges, which differ in their cavity width.31H NMR titrations in organic media have demonstrated that these macrocycles are able to bind several neutral organic guests bearing acidic methyl residues and that the rigidity of the ligand skeleton is an essential prerequisite for their efficiency.3b Arena et al. have carried out a detailed study of the inclusion of acetonitrile and nitromethane into some of these calix[4]arenes in CCl4.4 The thermodynamic parameters (ΔH° and ΔS°) indicate that the investigated receptors form complexes of comparable stability with acetonitrile and nitromethane, thus suggesting that the different guest acidity is not the only factor affecting the extent of binding.
Stibor et al. performed a 1H NMR study of the complexation of a series of neutral chloro, nitriles and nitro compounds with p-tert-butylcalix[4]arene derivatives, partially functionalised at the lower rim, in apolar solvents.5 The lack of correlation between the electron effect of the substituent at the lower rim of the host and the complex stability, led them to suggest that the binding properties are mainly controlled by the calix rigidity and by the ligand geometry. Arduini et al. have investigated the inclusion of a series of CH2XY guests of different acidity using partially alkylated p-tert-butylcalix[4]arene derivatives.6 This study indicates that the guest acidity cannot represent the sole factor responsible for the formation of the complexes. In fact, whereas, on one hand, a linear correlation between the extent of binding and the acidity of the CH2XY guests has been found, on the other hand, a dependence from the polarizability of the X and Y groups has also been shown.
Based on these results, the prerequisites, that determine the ability of cone conformers of calix[4]arenes to form endo-cavity inclusion complexes in apolar media, are: i. the presence in the guests molecule of acidic “activated” CH3 or CH2 groups and ii. the reduction of the conformational flexibility of the receptor. Specific CH–π(aromatic) interactions7 represent the main driving force for the formation of these complexes, as indicated by solution studies in apolar media and shown by structure data in the solid state. The analysis of the solid state structure of these calix[4]arene host–guest complexes indicate that, whereas CH3X guests are bound preferentially by hosts having an aromatic cavity with a C4v symmetry, CH2XY guests are bound in a complementary manner by calix[4]arene receptors, that can assume a flattened cone structure having a C2v symmetry.8
However no comparison of the selectivity of the recognition process in apolar solvents, using the two different approaches to the rigidification of the calixarene cone conformer, have been reported in the literature so far. In fact, whereas 1,3-dialkoxycalix[4]arene derivatives have been widely studied as receptors of several neutral species, biscrown-3-calix[4]arene derivatives have been employed as hosts only for CH3X guests.
In this paper we report a study of the binding of p-tert-butylcalix[4]arene cone conformers rigidified through different approaches. The comparatively more versatile‡ and more flexible 1,3-dipropoxy-p-tert-butylcalix[4]arene (1) and the more rigid biscrown-3-p-tert-butylcalix[4]arene (2) were selected as hosts (Fig. 1) for CH3X and CH2XY neutral guest molecules, to have information on the relative weight of host rigidity and guest complementarity in the recognition process. In previous studies it was demonstrated that, for guests bearing the methylene moiety, the binding process depends also upon the polarizability of the X and Y substituents; these studies also showed that the presence of CH3, Cl or CN groups did not markedly affect polarizability.6 Based on these findings, ClCH2CN, CH3CH2CN and CH2Cl2 were chosen as guests, to keep the polarizability of X and Y groups constant.
![Schematic representation of calix[4]arene derivatives 1 and 2.](/image/article/2004/NJ/b308996g/b308996g-f1.gif) | ||
| Fig. 1 Schematic representation of calix[4]arene derivatives 1 and 2. | ||
The systematic study of the energetics of the inclusion of CH3X and CH2XY guests into the π-donor cavity of calix[4]arene derivatives was carried out using 1H NMR spectroscopy in CCl4 and direct calorimetry. In fact, the determination of ΔH° and ΔS° contributions, which reveal information that are not expressed in the ΔG° term,9 may provide a better understanding of the forces involved in the inclusion processes and may help in explaining the different stability of the adducts formed. Further information was obtained by comparing the solid state structures of the adducts formed by hosts 1 and 2 with acetonitrile and chloroacetonitrile.
Results and discussion
NMR binding studies
In order to compare the impact of the different rigidification of hosts 1 and 2 on the formation of the adducts, the binding constant of biscrown-3-p-tert-butylcalix[4]arene (2) with ClCH2CN and CH3CH2CN were measured under the same experimental conditions used for host 1 (see Table 1).3,6 These two guests were chosen as suitable CH2XY type guests because of the similar polarizability of their X and Y groups.6 The spectroscopic titrations proved the formation of 1 ∶ 1 inclusion complexes in all cases.Moreover, the upfield shifts of the acidic CH protons indicated that each guest is included via its methylene moiety, thanks to CH–π interactions with the π-donor aromatic rings of the calixarene. The binding constant values were obtained by refining proton upfield shifts using a non-linear least-squares fitting procedures.10 On this base, considering CH3CN, and ClCH2CN as referring molecules of the two classes of guests, the relative binding constants in CCl4 were 150 and 102 M1 respectively for host (1) and 320 and 25 for host (2). As expected the less flexible host 2 turns out to be a more efficient and selective host for CH3X guests. On the contrary, the more flexible derivative 1 is able to recognise both classes of guests and shows a lesser selectivity for CH2XY guests.
Thermodynamic studies
Although in the previous studies6 the binding constants were determined either in CCl4 or in CDCl3, we only used CCl4 in the calorimetric investigations. This solvent, in fact, cannot form hydrogen bonds with the reactants and is also too big to fit into the p-tert-butylcalix[4]arene cavity. It is not surprising that the logK values (Table 2) are larger than those determined in chloroform (a competing guest). The choice of CCl4 was also dictated by the fast evaporation taking place when running the calorimetric experiment in chloroform, which hampered reproducible thermograms to be obtained (see experimental section).| Complex | logKd | logKe | ΔG°/kJ mol−1 | ΔH°/kJ mol−1 | TΔS°/kJ mol−1 |
|---|---|---|---|---|---|
| a This work; b see ref. 4; c standard deviation values are given in parentheses; d logK values determined via1H NMR; e logK values determined by direct calorimetry. | |||||
1
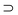 CH3CN CH3CN |
2.18 | 2.23(3) | −12.73 | −35.2(6) | −22.5(7) |
|
1
ClCH2CN |
2.01 | 1.95(4) | −11.13 | −39.1(3) | −28.0(3) |
|
1
CH3CH2CN |
1.48 | 1.43(5) | −8.16 | −27.7(4) | −19.6(4) |
|
1
CH2Cl2 |
0.92 | 0.99(5) | −5.65 | −23.1(4) | −17.5(3) |
|
2
CH3CN |
2.5 | 2.39(5) | −13.64 | −39.3 | −25.4 |
Fig. 2 shows the total net heat (i.e. corrected for the dilution) for the reaction of CH3CN with host 1vs. the volume of titrant added. The guest/host ratio, at the end of the reaction (see ESI†), was set for each system in such a way as to reach saturation; only in this condition, K and ΔH° can be determined simultaneously. The results of the calorimetric study are reported in Table 2. The logK values determined calorimetrically are in excellent agreement, within the experimental error, with the figures obtained from 1H NMR titrations (see Table 2). The ΔG° values indicate that the stability of the complexes with CH2XY guests parallels their acidity (ClCH2CN,11 pKa-DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =26; CH3CH2CN,11 pKa-DMSO=31; CH2Cl2,11 pKa-DMSO=35), while the complex with CH3CN is the most stable among the investigated adducts.
=26; CH3CH2CN,11 pKa-DMSO=31; CH2Cl2,11 pKa-DMSO=35), while the complex with CH3CN is the most stable among the investigated adducts.
![Calorimetric run for the titration of 25 ml of 1,3-dipropoxy-p-tert-butylcalix[4]arene (1)
(0.00144 mol dm−3) with CH3CN (0.53203 mol dm−3) in CCl4.](/image/article/2004/NJ/b308996g/b308996g-f2.gif) | ||
| Fig. 2 Calorimetric run for the titration of 25 ml of 1,3-dipropoxy-p-tert-butylcalix[4]arene (1) (0.00144 mol dm−3) with CH3CN (0.53203 mol dm−3) in CCl4. | ||
The dissection of the ΔG° values into the enthalpic and the entropic terms provides further insight into the forces driving the recognition processes and reveals some details that are not expressed by the logK values. The thermodynamic data (ΔH° and ΔS°) show that inclusion process is enthalpically driven for all systems, whilst the entropy of binding is unfavourable in all cases, as expected for the interaction of neutral partners in organic media. Table 2 shows that the binding efficiency of 1 is directly correlated with the enthalpic contribution. CH3CN and ClCH2CN complexes, which have the largest binding constants, are also characterised by the largest enthalpic contributions.
For the CH2XY guests, i.e. for ClCH2CN, CH3CH2CN and CH2Cl2 complexes, the stabilities are linearly correlated with the acidity of CH2 groups (ClCH2CN,11 pKa-DMSO=26; CH3CH2CN,11 pKa-DMSO=31; CH2Cl2,11 pKa-DMSO=35). A higher acidity of the aliphatic moieties results also in a larger ΔH° of binding for the corresponding adduct and, ultimately, in a larger ΔG°. This indicate that CH–π interactions, that are maximized in the presence of acid methylene residues, are the forces principally responsible for the formation of this class of complexes in organic media. The entropic term is significantly negative for all the systems investigated (see Table 2), due to reduction of degrees of freedom resulting from complex formation. As shown previously, p-tert-butyl functionalities widen and deepen the typical calix cavity, thus increasing the interaction points. This, leads on the one hand to a more favourable enthalpic contribution, but on the other causes a more pronounced stiffening of the host–guest system. In line with this reasoning, the more negative ΔS° values are obtained for the systems presenting the higher enthalpic stabilization, as the complexes formed with CH3CN and ClCH2CN. The CH3CN complex is the most stable among the systems investigated in this study. For the sake of comparison, in Table 2 we have also reported the thermodynamic parameters for the inclusion of CH3CN in host 2. The inclusion of CH3CN into this more preorganized and rigid host is more stabilized (ΔG°: −13.64 vs.
−12.73 kJ mol−1) than the inclusion in host 1. The residual mobility of host 1 worsens the efficiency of the receptor; the rigid host 2, blocked in the cone conformation (a C4v symmetry), can have a larger number of interactions with CH3X guests and turns out to be more efficient in their recognition. This extra-stabilization mainly derives from a more favourable enthalpic contribution and it seems that the different rigidification approach used in the two receptors is not reflected by the corresponding variation of the entropic contribution. However, differently from what is observed for CH2XY guests, for CH3CN the binding constant is not straightforwardly correlated either with the guest acidity (CH3CN,12 pKa-DMSO=31.3; ClCH2CN,11 pKa-DMSO=26) or with the enthalpic contribution.
As previously hypothesized by Arduini et al.,6 the larger binding constant value observed for CH3CN, and more generally for CH3X complexes, is the result of the incorporation of the guest methyl group that, upon inclusion into the cavity of 1, still maintains its rotational freedom along its C–C or C–X bond. This implies a smaller loss of mobility in the formation of the [1CH3–X] if compared to the complexation of the more mobile CH2XY guests. The thermodynamic parameters (ΔH and ΔS) support this hypothesis. In fact, if we compare CH3CN and ClCH2CN complexes with ligand 1, which are those described by the larger logK values, we can notice that the higher stability of the [1CH3CN] complex does not result from a more favourable enthalpic term, but from a less unfavourable entropic contribution. This also indicates that the acidity of CH groups is not the sole factor affecting the extent of binding of CH3X guests. Moreover the different geometry of the complexes and the difference in the host–guest contact do not allow a direct comparison of the interactions present in the complexes with CH3–X and CH2XY guests, respectively.
Solid state structural studies
Further information on the recognition processes of CH3CN and ClCH2CN guests were obtained by comparing the solid state structures of their complexes with hosts 1 and 2.The solid state complexes of host 1 with acetonitrile and host 2 with acetonitrile and chloroacetonitrile were prepared. Their structures were determined by X-ray diffractometric methods and compared with those previously resolved for host 1 and chloroacetonitrile.6 The molecular structures of the four complexes are shown in Fig. 3. These structures were analysed to understand the nature and the strength of the host–guest interactions. In this analysis we have taken advantage of the use of appropriate geometrical descriptors,8 in order to have a rational base to describe the host geometries and the orientations of the guests inside the host cavities (see Fig. 4). The calculated values are reported in Table 3.
 | ||
| Fig. 3 Top and side views of the X-ray solid state inclusion complexes of a)
1CH3CN, b)
2CH3CN, c)
2ClCH2CN and d)
1ClCH2CN.6 | ||
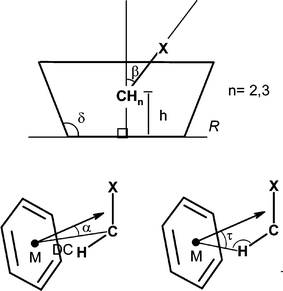 | ||
| Fig. 4 Geometrical descriptors for solid state host–guest complexes.8 | ||
| Complex | δ 1 [°] | δ 2 [°] | δ 3 [°] | δ 4 [°] | β [°] | h [Å] | DC1[Å] | DC2[Å] | DC3[Å] | DC4[Å] | α 1 [°] | α 2 [°] | α 3 [°] | α 4 [°] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Calculated from CN. b Disordered guest : distances h calculated from Cl atom are 2.843(9), 2.426(7) and 2.462(7) Å. | ||||||||||||||
| 1⊂CH3CN | 116.53(4) | 117.54(4) | 116.33(4) | 117.41(4) | 16.44(7) | 2.699(2) | 3.535(3) | 3.713(3) | 3.545(3) | 3.699(3) | 3.77(5) | 5.46(5) | 4.49(5) | 4.80(5) |
| 2⊂CH3CN | 113.9(1) | 115.1(1) | 118.0(1) | 114.6(1) | 5.9(2) | 2.814(6) | 3.681(8) | 3.655(6) | 3.656(7) | 3.632(8) | 1.4(1) | 1.2(1) | 1.7(1) | 1.1(1) |
| 2⊂ClCH2CN | 115.5(2) | 118.0(1) | 117.4(1) | 116.1(2) | 5.9(2)a | b | b | b | b | b | b | b | b | b |
| 1⊂ClCH2CN | 110.3(2) | 138.2(2) | 109.7(2) | 135.3(2) | 48.5(4) | 2.857(2) | 3.548(3) | 3.934(3) | 3.552(3) | 4.103(3) | 6.06(5) | 15.81(5) | 6.24(6) | 19.04(5) |
The structure refined for 1⊂CH3CN (see Fig. 3a) shows that the shape of this complex is mainly determined by two strong intramolecular hydrogen bonds which link the OH of one unfunctionalised phenolic ring to the phenolic oxygen of the adjacent functionalised rings [D⋯A distances: 2.768(2) and 2.748(2)
Å; D–H⋯A angles: 176(2) and 172(3)°]. This is significantly different from what found for the corresponding chloroacetonitrile complex, where the analogous hydrogen bonds are stronger [D⋯A distances: 2.712(2) and 2.675(2)
Å; D–H⋯A angles: 171(3) and 169(2)°].
However it is noteworthy that the host is not completely rigid since a little change in the geometrical parameters of the two (although strong) intramolecular hydrogen bonds (obtainable with little expense in enthalpy) can induce a significant conformational change at the upper-rim. Consequently, the host can change the symmetry of its aromatic cavity for a better steric match of the guest. The δ1–4 values reported in Table 3 show that the host cavity turns into an almost flattened partial cone C2v-like or into a C4v-like symmetry to better fit CH2ClCN (C2v symmetry) or the cylinder-like CH3CN guest, respectively.
Unlike 1, the more rigid biscrown-3-p-tert-butylcalix[4]arene 2 lacks this ability. In fact, in the two CH2ClCN and CH3CN complexes, the host cavity remains in an almost C4v symmetry and is unaffected by the guest shape.
Therefore the aromatic cavity can host CH3CN by including its methyl group, but it is not flexible enough to host CH2ClCN by including its CH2 group. As a consequence the latter guest enters in the cavity by inserting its chlorine atom and this geometry weakens the CH–π intermolecular host–guest attractive interaction in the solid state.
Both hosts 1 and 2 show a pseudoC4 symmetry in the two complexes with acetonitrile (see δ angles in Table 3); the more mobile 1 is slightly more symmetrical than host 2. In both complexes, the methyl group is included into the aromatic cavity of the hosts; however, the methyl group of the guest is not perfectly perpendicular to the plane defined by the methylene groups of the calixarene (see β angle in Table 3 and Fig. 4) and the deviation from the perpendicularity is greater in the complex with host 1. Moreover, the geometrical descriptors for the CH3CN complex with 1 and 2 show that in the former adduct the host skeleton is more open and consequently the guest molecule is more deeply buried in the cavity. In fact, the distance h of the CMethyl from the weighted least-squares plane through the four bridging methyl groups of the calix[4]arene are 2.699(2) and 2.814(6) Å, respectively.
CH3CN and 2CH3CN complexes makes it possible to compare host–guest interactions in these two complexes. In both the cases, the guest is linked to the host via a couple of weak intermolecular CH–π interactions whose geometrical parameters are summarized in Table 4.
| C–H⋯Mb | H⋯M/Å | C–H⋯M/° | C⋯M/Å | τ/°c | |
|---|---|---|---|---|---|
| a See Figs. 3a and 3d for ring and guest tags. b M is the ring centroid. c τ is the angle formed by H⋯M line and the normal to the ring (→), see Fig. 4. | |||||
| 1 ⊂ CH3CN | C–H3G⋯B | 2.90(5) | 142(4) | 3.713(3) | 14.5(8) |
| C–H2G⋯D | 2.87(5) | 149(4) | 3.669(3) | 3.1(9) | |
| 2 ⊂ CH3CN | C–H1G3⋯A | 2.79(1) | 153(1) | 3.681(8) | 7.3(1) |
| C–H1G⋯C | 2.70(1) | 164(1) | 3.656(7) | 15.7(1) | |
| 1 ⊂ ClCH2CN | C–H1G2⋯A | 2.624(2) | 161.4(2) | 3.548(3) | 4.61(4) |
| C–H1G1⋯C | 2.669(2) | 167.6(2) | 3.552(2) | 2.98(5) | |
The four equilibrium distances CGuest⋯M (M=ring centroid) indicate that the guests are stabilized by CH–π interactions. Tzuzuki et al. reported the most accurate calculation of the magnitude of the CH–π interactions obtained so far for the methane–benzene synthon.13 They found an equilibrium distance of 3.8 Å and the binding energy was 1.45 kcal mol−1. For the more “acidic” CH in CH3Cl interacting with benzene, the equilibrium distance decreases to 3.6 Å and the binding energy goes up to 3.0 kcal mol−1, but the energy profile is large around the minimum so that considerably attraction still exists up to 3.8 Å.7b
In the two acetonitrile complexes here reported all the four CGuest⋯M are close to 3.6 rather than to 3.8 Å and the angles C–H⋯M and τ (see Fig. 4) indicate that in both complexes the C–HMethyl bond is almost aligned to the normal to the ring (through its centroid) as expected for an ideal CH–π interaction geometry.
ClCH2CN. All the calculated geometrical parameters indicate that the CH–π interactions are the strongest ones among the series; in fact, the H⋯M and C⋯M distances and the C–H⋯M angle are the shortest and the greatest, respectively, among the series of complexes investigated.
In the 2CH2ClCN complex, the aromatic cavity is occupied by the chlorine atom and the guest is disordered. This structure suggests that dispersive interactions are involved in the stabilization of this complex and this would explain the low binding constant observed in apolar solvents.
Conclusions
This multidisciplinary approach to the study of the recognition of guests containing CH3 and CH2 acidic groups by differently rigidified cone conformers of p-tert-butylcalix[4]arene provides new interesting information on these processes. In particular the rigidity of host 2 and the different complexes with the two types of guests can be easily explained on the basis of the different structures of the complexes. The thermodynamic data evidenced that these processes are enthalpically driven but that it is the entropy difference that favours the recognition of CH3X guests. CH–π interaction, evidenced by solid state studies, explain the enthalpic data and in particular the better efficiency of host 2 in the recognition process of acetonitrile.Experimental
Acetonitrile, dichloromethane and NaCl (Merck, >99.5%) chloroacetonitrile, propionitrile and 18-crown-6 (Aldrich, 99%) were commercial products and were used without any further purification. Carbon tetrachloride and methanol (>99.8%) were purchased from Carlo Erba. Calix[4]arene derivatives (1)14 and (2)3b were synthesized analogously to the procedures previously described.NMR experiments
1H NMR titration experiments were performed adding increasing amounts of a 0.5–1.0×10−1 M stock solution of the guest to a 0.5–1.0×10−2 M stock solution of the host in CCl4(C6D6 as external standard), by monitoring the guest chemical shift variation. All the NMR spectra showed time-averaged signals for the free and complexed species and, having verified a 1 ∶ 1 stoichiometry for the host–guest association by means of continuous variation methods,15 the stability constants (K) for the complex formation were calculated using methods that have been previously described based on the non-linear fitting of the experimental data.10
Calorimetric measurements
The calorimetric titrations were performed at 25.000±0.001°C in CCl4, using a Tronac 450 (Utah, USA) isoperibolic calorimeter. This calorimeter measures the temperature changes, following the addition of titrant, through a precision thermistor, which generates a voltage output; this output is converted into a heat quantity by a precision heater. This instrument was equipped with a 25 ml Dewar cell, although this dramatically increased the amount of calix[4]arene needed for each titration, since fast evaporation phenomena were observed employing the 4 ml Dewar.4 Otherwise, regular and reproducible baselines of the thermograms were obtained with the 25 ml Dewar cell, with a marked positive slope. Usually a 0.5–1.5 mol dm−3 solution of the guest (CH3CN, ClCH2CN, CH3CH2CN or CH2Cl2) was added to a 1.2–1.5×10−3 mol dm−3 solution of host 1, recording 30–40 points for each of the 4–6 independent titrations, maintaining the guest–host molar ratios at the end of the titration in the range of 15.2–27.0 (see ESI†). Blank experiments were carried out before each titration, in order to correct the experimental data for all non chemical energy contributions, such as the heat of dilution and heat of friction effects, resulting from the addition of the titrant to the titrate solution in the calorimetric vessel. The calorimeter was calibrated electrically (twice for each single run, before and after the titration) to check for accuracy and reproducibility. The calorimeter was calibrated chemically too, by titrating a solution of 18-crown-6 in methanol with a methanolic solution of NaCl. The ΔH° and logK values were in good agreement with the literature values.16 The experimental data were treated by using a modified version of the computer program EQDH,17 able to perform a simultaneous refinement of logK and ΔH° values.
X-ray data collection, structure solution, and refinement
Crystal data end experimental detail for the three structures are collected in Table 5. The raw frame data of 2ClCH2CN were processed using SAINT18 and SADABS19 to yield the reflection data file.
CH3CN, 2CH3CN, and 2ClCH2CN inclusion compounds
|
a
F
0≥4σ(F0).
b
R
1=Σ
‖
F0|−|
Fc‖/
Σ
|F0|, wR2=[Σ w(F20−F2c)2/Σw F40]1/2. Goodness-of-fit=[Σ w(F20−F2c)2/(n−p)]1/2, where n is the number of reflections and p the number of parameters.
|
|||
|---|---|---|---|
| Compound |
1
CH3CN |
2
CH3CN |
2
ClCH2CN |
| Molecular formula | C52H71NO4 | C54H71NO6 | C54H70ClNO6 |
| Formula weight | 774.137 | 830.158 | 864.603 |
| Crystal system | Monoclinic | Triclinic | Monoclinic |
| Space group | P21/a |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21 |
| a [Å] | 19.892(5) | 12.064(5) | 11.237(5) |
| b [Å] | 19.296(5) | 19.817(5) | 19.743(5) |
| c [Å] | 12.102(5) | 11.242(5) | 12.286(5) |
| α [°] | 90 | 90.27(2) | 90 |
| β [°] | 90.69(2) | 114.16(2) | 113.69(2) |
| γ [°] | 90 | 88.53(2) | 90 |
| V [Å3] | 4645(3) | 2451(2) | 2496(2) |
| Z | 4 | 2 | 2 |
| ρ (calcd.) [g cm−3] | 1.107 | 1.125 | 1.150 |
| F(000) | 1688 | 900 | 932 |
| T [K] | 173 | 298 | 298 |
| λ [Å] | 1.54178 | 1.54178 | 0.71069 |
| μ [mm−1] | 5.262 | 5.628 | 1.248 |
| Reflections collected | 9340 | 9264 | 26879 |
| Independent refections | 8620 | 9264 | 10604 |
| R int | 0.013 | 0.000 | 0.025 |
| Observed reflectionsa | 7380 | 1957 | 5717 |
| Data/parameters/ restraints | 551/0 | 549/0 | 578/9 |
| Goodness-of-fit on F2b | 1.472 | 0.832 | 1.116 |
| R 1 | 0.0505 | 0.0765 | 0.0977 |
| wR 2 | 0.1818 | 0.1980 | 0.3275 |
| Largest diff. peak and hole [e Å−3] | 0.57, −0.36 | 0.46, −0.23 | 0.70, −0.81 |
The three structures were solved by direct methods with SIR9220 and refined with SHELXL-97.21 In the structure of 2CH3CN the tert-butyl groups were affected by severe static disorder. In 2ClCH2CN the guest showed severe static disorder with the C and Cl atoms of the terminal CH2Cl group statistically distributed over two and tree different positions respectively. Molecular geometries were analysed with PARST97.22,§
Acknowledgements
Progetti di Ricerca di Ateneo – Università degli Studi di Catania – anno 2002 and Cofin 2002 (anno 2002-prot. 2002038825_006), MIUR “Supramolecular Devices Project” and I.N.C.A. are gratefully acknowledged for financial support. We are grateful to CIM (Centro Interdipartimentale di Misure) “G. Casnati” of the University of Parma for NMR and Mass measurements. The authors warmly thank Mrs Valeria Zito (Istituto Biostrutture e Bioimmagini Sez. di Catania CNR) for technical support.References
- For comprehensive reviews on calixarenes see: (a) V. Böhmer, Angew. Chem., Int. Ed. Engl., 1995, 34, 713 CrossRef; (b) C. D. Gutsche, Calixarenes Revisited, Royal Society of Chemistry, Cambridge, 1998 Search PubMed; (c) Z. Asfari, V. Böhmer, J. Harrowfield, and J. Vicens, Calixarenes 2001, Kluwer Academic Publishers, Dordrecht, Boston, London, 2001 Search PubMed; (d) Calixarenes in Action, eds. L. Mandolini, R. Ungaro, Imperial College Press, London, 2000 Search PubMed.
- (a) Z. Asfari, V. Böhmer, J. Harrowfield, and J. Vicens, Calixarenes 2001, Kluwer Academic Publishers, Dordrecht, Boston, London, 2001, pp. 440–456 Search PubMed; (b) G. Arena, A. Casnati, A. Contino, D. Sciotto and R. Ungaro, Tetrahedron Lett., 1997, 36, 4685 CrossRef CAS.
- (a) A. Arduini, M. Fabbi, M. Mantovani, L. Mirone, A. Pochini, A. Secchi and R. Ungaro, J. Org. Chem., 1995, 60, 1454 CrossRef CAS; (b) A. Arduini, W. M. McGregor, D. Paganuzzi, A. Pochini, A. Secchi, F. Ugozzoli and R. Ungaro, J. Chem. Soc., Perkin Trans. 2, 1996, 839 RSC.
- G. Arena, A. Contino, A. Magrì, D. Sciotto, A. Arduini, A. Pochini and A. Secchi, Supramol. Chem., 2001, 13, 379 CAS.
- (a) S. Smirnov, V. Sidorov, E. Pinkhassik, J. Havlicek and I. Stibor, Supramol. Chem., 1997, 8, 187 CAS; (b) P. Lhoták, R. Zieba, V. Hromádko, I. Stibor and J. Sykora, Tetrahedron, 2003, 44, 4519 CAS.
- A. Arduini, G. Giorgi, A. Pochini, A. Secchi and F. Ugozzoli, Tetrahedron, 2001, 57, 2411 CrossRef CAS.
- (a) F. Ugozzoli, A. Arduini, C. Massera, A. Pochini and A. Secchi, New. J. Chem., 2002, 26, 1718 RSC; (b) S. Tsuzuki, K. Honda, T. Uchimaru, M. Mikami and K. Tanabe, J. Phys. Chem. A, 2002, 106, 4423 CrossRef CAS; (c) M. Nishio, M. Hirota and Y. Umezawa, The CH/π interaction, Wiley-VCH, NY, 1998 Search PubMed; (d) O. Takahashi, Y. Kohno, S. Iwasaki, K. Saito, M. Iwaoka, S. Tomoda, Y. Umezawa, S. Tsuboyama and M. Nishio, Bull. Chem. Soc. Jpn., 2001, 74, 2421 CrossRef CAS; (e) O. Takahashi, S. Tsuboyama, Y. Umezawa, K. Honda and M. Nishio, Tetrahedron, 2000, 56, 6185 CrossRef CAS; (f) Y. Umezawa, S. Tsuboyama, H. Takahashi, J. Uzawa and M. Nishio, Bioorg. Med. Chem., 1999, 7, 2021 CrossRef CAS; (g) T. Steienr and G. R. Desiraju, Chem. Commun., 1998, 891 RSC; (h) D. H. Williams and M. S. Westwell, Chem. Soc. Rev., 1998, 27, 57 RSC; (i) R. E. Grillard, F. M. Raymo and J. F. Stoddart, Chem. Eur. J., 1997, 3, 1933 CrossRef CAS; (j) For a comprehensive literature list see http://www.tim.hi-ho.ne.jp/dionisio/.
- A. Arduini, F. F. Nachtigall, A. Pochini, A. Secchi and F. Ugozzoli, Supramol. Chem., 2000, 12, 273 CAS.
- (a) G. Arena, A. Contino, T. Fujimoto, D. Sciotto and Y. Aoyama, Supramol. Chem., 2000, 11, 279 CAS; (b) G. Arena, A. Casnati, A. Contino, G. Lombardo, D. Sciotto and R. Ungaro, Chem. Eur. J., 1999, 5, 738 CrossRef CAS.
- (a) C. S. Wilcox, in Frontiers of Supramolecular Organic Chemistry and Photochemistry, eds. H.-J. Schneider, H. Dürr, VCH, Weinheim, 1991, pp. 123–143 Search PubMed; (b) K. A. Connors, Binding Constants, Wiley, New York, 1987 Search PubMed; (c) For a recent review on determination of association constants using NMR data, see: L. Fielding, Tetrahedron, 2000, 56, 6151 Search PubMed and references therein.
- K. Yoshimura and Y. Fukazawa, Tetrahedron Lett., 1996, 37, 1435 CrossRef CAS.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456 CrossRef CAS.
- S. Tzuzuki, K. Honda, T. Uchimaru, M. Mikami and K. Tanabe, J. Am. Chem. Soc., 2000, 122, 3746 CrossRef CAS.
- J. van Loon, A. Arduini, L. Coppi, W. Verboom, A. Pochini, R. Ungaro, S. Harkema and D. N. Reinhoudt, J. Org. Chem., 1990, 55, 5639 CrossRef CAS.
- P. Job, Ann. Chim., 1928, 9, 113 CAS.
- R. M. Izatt, R. E. Terry, B. L. Haymore, L. D. Hansen, N. K. Dalley, A. G. Avondet and J. J. Christensen, J. Am. Chem. Soc., 1976, 98, 7620 CrossRef CAS.
- E. A. Eatough, J. J. Christensen and R. M. Izatt, Thermochimica Acta, 1972, 3, 219 CrossRef CAS.
- SAINT Software Users Guide, Version 6.0, Bruker Analytical X-ray Systems, Madison, WI, 1999.
- G. M. Sheldrick, SADABS, Bruker Analytical X-ray Systems, Madison, WI, 1999.
- A. Altomare, M. C. Burla, M. Camalli, G. Cascarano, C. Giacovazzo, A. Guagliardi, G. Polidori, SIR92, J. App. Crystallogr., 1994, 27, 435 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997http://shelx.uni-ac.gwdg.de/shelx/index.html Search PubMed.
- M. Nardelli, PARST97, updated version of PARST95, J. Appl. Crystallogr., 1995, 28, 659 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: experimental conditions used for calorimetric measurements. See http://www.rsc.org/suppdata/nj/b3/b308996g/ |
| ‡ 1,3-Disubstituited calix[4]arene derivatives represent a versatile class of synthetic receptors. In fact, this class of compounds makes it easy to realise active components of new artificial sensors for small organic molecules based on calix[4]arene compounds having 1,3-dialkoxy substituents functionalised with groups that allow the anchoring of the host onto sensor surfaces. |
| § CCDC reference numbers 215593 (2CH3CN), 215594 (2ClCH2CN), 215595 (1CH3CN). See http://www.rsc.org/suppdata/nj/b3/b308996g/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2004 |