CoCl2 catalysed decarboxylation-oxidation of mandelic acids by molecular oxygen
Isabelle
Favier
a,
Elisabet
Duñach
*ab,
Dominique
Hébrault
c and
Jean-Roger
Desmurs
d
aLaboratoire Arômes, Synthèses et Interactions, Université de Nice-Sophia Antipolis, Parc Valrose, 06108, Nice cedex 2, France
bLaboratoire Chimie Bioorganique (CNRS UMR 6001), Université de Nice-Sophia Antipolis, Parc Valrose, 06108, Nice cedex 2, France
cRhodia Organique Fine, Centre de Recherches, BP 62, 85, Av. Frères Perret, 69192, Saint Fons cedex, France
dRhodia Organique Fine, 190, Av. Thiers, 69457, Lyon cedex 06, France
First published on 29th October 2003
Abstract
A series of mandelic acid derivatives was oxidised by molecular oxygen using cobalt(II) chloride as the catalyst. Benzaldehyde and/or benzoic acid derivatives were obtained in high selectivities, depending on the aromatic ring substitution. Different oxidation mechanisms are operating, depending on the mandelic acid substitution.
Introduction
Cobalt complexes, and in particular cobalt(II) chloride, constitute versatile catalysts in organic synthesis1 and have been used, for example, in the monohydroxylation of β-keto esters,2 for the synthesis of β-amino alcohols from oxiranes3 and for the acylation of anisole.4In combination with molecular oxygen, the oxidation of alkenes to epoxides with cobalt complexes has been described with Schiff base ligands, cryptates or porphyrins.5–8 Under O2, the formation of cobalt(III) superoxo species, responsible for the oxidation of the alkenes, has been proposed. The oxidative cleavage of alkenes versus their epoxidation by a Co(II)/O2 system has been studied for the cleavage of olefins into carbonyl derivatives9 and for the oxidative cleavage of isoeugenol to vanillin.10 The oxidative cleavage of α-diols has also been reported.11 The same system has also been applied for the oxidation of alcohols to the corresponding carbonyl compounds,7 for the oxidation of alkylbenzenes to carboxylic acids12 and the oxidation of cyclic ethers to lactones.13
The oxidation of mandelic acids has been examined under a variety of systems.14 However, the Co-catalysed oxidative decarboxylation of mandelic acid derivatives has not yet been reported. We present here our results on the use of the CoCl2/O2 system for the oxidation of such substrates. Moreover, we recently examined the use of Bi(0)/O2 for the oxidation of α-ketols, and epoxides,15 as well as for the oxidation of mandelic acids.16,17 Some comparisons between both catalytic systems are presented.
Results and discussion
Oxidation of mandelic acid derivatives catalysed by CoCl2/O2
The oxidation of several mandelic acid derivatives (1) was examined under different reaction conditions, using CoCl2 as the catalyst. As illustrated in Scheme 1, the oxidative cleavage of mandelic acids mainly affords benzoic acid derivatives (2) and/or benzaldehyde derivatives (3).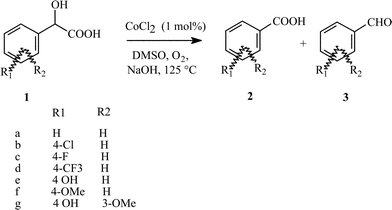 | ||
| Scheme 1 Oxidation reaction of mandelic acid derivatives showing the numbering scheme | ||
In a water medium at 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C, the conversion of 1a was less than 5% after 24 h and in H2O–NaOH or H2O–AcOH media the conversion of 1a was less than 18%. The best results were obtained in DMSO at 125°C with 1 mol % catalyst under atmospheric O2 pressure. It was observed that the oxidation reaction could be optimised by operating under basic conditions; the addition of 1.5 equiv. (versus substrate) of a 50% aqueous solution of sodium hydroxide afforded the best yields, as compared to the reaction in DMSO alone or with AcOH as the additive. Both DMSO and NaOH seem to play an important role in accelerating the oxidation. The results of the oxidation of several mandelic acid derivatives (1) under O2
(1 atm) in a cobalt(II) chloride catalysed reaction are presented in Table 1.
°C, the conversion of 1a was less than 5% after 24 h and in H2O–NaOH or H2O–AcOH media the conversion of 1a was less than 18%. The best results were obtained in DMSO at 125°C with 1 mol % catalyst under atmospheric O2 pressure. It was observed that the oxidation reaction could be optimised by operating under basic conditions; the addition of 1.5 equiv. (versus substrate) of a 50% aqueous solution of sodium hydroxide afforded the best yields, as compared to the reaction in DMSO alone or with AcOH as the additive. Both DMSO and NaOH seem to play an important role in accelerating the oxidation. The results of the oxidation of several mandelic acid derivatives (1) under O2
(1 atm) in a cobalt(II) chloride catalysed reaction are presented in Table 1.
°C
Unsubstituted mandelic acid (1a) reacted smoothly, with a conversion of 42% after 24 h. No important evolution of the system was observed with longer reaction times. A 89∶11 mixture of 2a and 3a was formed in 62% yield. The remaining product was phenylglyoxylic acid (4a), obtained in 38% yield.
Interestingly, better conversions were obtained with substituted mandelic acids, either with electron-donating or with electron-withdrawing substituents. Thus, the reactivity of p-chloro-, p-fluoro- and p-trifluoromethylmandelic acids (1b–1d) possessing electron-withdrawing substituents was examined; these substrates afforded conversions of 75–100% after 24 h. A clean reaction with a quantitative yield of the corresponding benzoic acids (2b–2d) and benzaldehydes (3b–3d) was obtained. However, the selectivity 2∶3 was very dependent on the nature of the substituents. Thus, for p-fluoro- and p-trifluoromethylmandelic acids (1c and 1d) the corresponding benzoic acid derivatives 2c and 2d were formed as the major compounds in 82–83% selectivities. In contrast, for p-chloromandelic acid (1b) the main product was p-chlorobenzaldehyde (3b) with a selectivity of 71%.
The reactivity was enhanced with electron-rich substituents (1e–1g). p-Hydroxymandelic acid (1e) was converted in 0.5 h and vanillic acid (1g) in 2 h. Yields of 2+3 of 57–88% was accounted for by the presence of some polymeric material in the cases of 1e and 1g and of an additional ketoacid derivative (4f, 12%), in the case of 1f. The selectivities 2∶3 were here in favour of the formation of the corresponding aldehydes 3e–3g, with 2∶3 ratios up to 1:99 in the cases of 4-hydroxymandelic acid (1e) and vanillic acid (1g). In the case of the p-methoxy substituent in 1f, the selectivity towards the aldehyde 3f was 84%.
The kinetics of the consumption of different mandelic acid derivatives is presented in Fig. 1
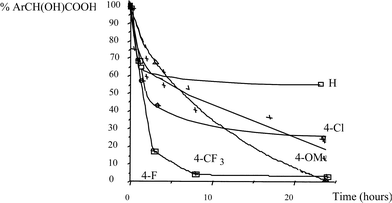 | ||
| Fig. 1 Kinetics of the oxidation of mandelic acid derivatives by the catalytic system CoCl2/O2 in DMSO–NaOH at 125 °C | ||
Mechanistic considerations
The reactivity and selectivity differences observed for the oxidation of mandelic acid derivatives are probably due to different mechanisms operating in the oxidative Co catalysed system. The oxidation of mandelic acids to the corresponding benzoic acids and/or benzaldehyde derivatives involves an oxidative decarboxylation reaction, which can occur via paths “a” or “b” shown in Scheme 2. Thus, derivatives 1 can be oxidised to the corresponding α-ketoacids 4via path “a”, with further decarboxylation to aldehydes 3 or oxidative decarboxylation to benzoic acids 2.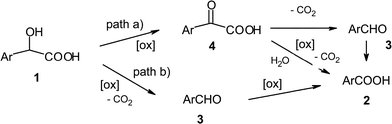 | ||
| Scheme 2 Mechanistic pathways for the oxidation of mandelic acids | ||
Alternatively, following path “b”, the oxidative decarboxylation of 1 can directly afford aldehydes 3, which may be further oxidised to acids 2. In order to determine the main reaction pathways for the differently substituted mandelic acid derivatives, mechanistic studies were carried out on the oxidation of substrates 1, as well as on the oxidation of the corresponding benzaldehydes 3 and ketoacid derivatives 4, catalysed by CoCl2. These reactions were carried out for unsubstituted 1a, for the mandelic acid derivative 1f with an electron-rich aryl ring and for 1d, with an electron-poor aryl moiety.
°C, unsubstituted mandelic acid (1a) was 35% converted after 2 h to yield benzoic acid (2a) and phenylglyoxylic acid (4a) with a selectivity of 57∶43. After 24 h, the conversion of 1a was of 42% and some benzaldehyde (3a) was present, with a 2a∶3a∶4a selectivity of 55∶7∶38 (Scheme 3).
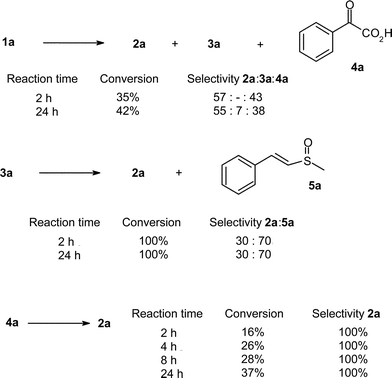 | ||
| Scheme 3 Oxidation of mandelic acid (1a), benzaldehyde (3a) and phenylglyoxylic acid (4a) catalysed by CoCl2 | ||
Both benzaldehyde (3a) and ketoacid (4a), possible intermediates in the formation of benzoic acid (2a), were subjected to oxidation under the same conditions. The oxidation of 3a afforded a complete conversion after 2 h, with the formation of 30% of 2a and 70% of a sulfoxide derivative (5a) issued from the reaction of 3a and DMSO. However, compound 5a was not isolated during the oxidation of 1a.
The oxidation of ketoacid 4a occurred at a slow rate and gave a conversion of only 16% after 2 h, with the exclusive formation of 2a. The conversion of 4a reached 37% after 24 h. The slow oxidation of 4a is in agreement with the fact it was isolated in 38–43% selectivity from the oxidation of 1a.
Taking into consideration the data of Scheme 3 and the fact that the sulfoxide derivative 5a was not formed during the oxidation of 1a, the mechanism of formation of 2a from 1a by the CoCl2 system can occur through the two different pathways “a” and “b” of Scheme 2. The main pathway ”b” involves a first oxidation of 1a to aldehyde 3a, followed by its further and rapid oxidation into 2a. Surprisingly, in this oxidation process from 1a compound 5a is not observed. Possibly the rapid oxidation of the intermediate aldehyde species prevents the formation of 5a.
In competitive pathway “a”, phenylglyoxylic acid (4a) is formed and accumulated and is only slowly oxidised into 2a. This pathway “a” is to be considered as a minor contribution, as illustrated in Scheme 4.
 | ||
| Scheme 4 Mechanistic pathways for the oxidation of mandelic acid (1a) catalysed by CoCl2 | ||
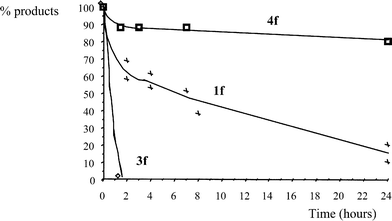 | ||
| Fig. 2 Oxidation of 4-methoxymandelic acid (1f), 4-anisaldehyde (3f) and 4-methoxyphenylglyoxylic acid (4f) catalysed by CoCl2 | ||
Table 2 presents the results obtained for the oxidations of 1f, 3f and 4f catalysed by CoCl2. As already observed for the oxidation of 3a, the oxidation of 4-anisaldehyde (3f), which was completely converted in 2 h, afforded mainly the sulfoxide derivative 5f (analogous to 5a in Scheme 3) in 90% yield while 4-methoxybenzoic acid (2f) was only formed in 10% yield. The sulfoxide derivative 5f was not observed during the oxidation of 1f.
| Substrate | Time/h | % Conversion | Products | % Product yield |
|---|---|---|---|---|
| 3f | 2 | 100 | 2f | 10 |
| 5f | 90 | |||
| 4f | 24 | 20 | 2f | 100 |
| 1f | 8 | 62 | 2f | 14 |
| 3f | 74 | |||
| 4f | 12 |
Our data of Fig. 2 and Table 2 indicate that the CoCl2 catalysed oxidation of 4-methoxymandelic acid (1f) should mainly proceed through mechanistic pathway “b” in Scheme 2, involving the direct formation of 4-anisaldehyde (3f). The carboxylic acid 2f should be formed from the oxidation of 3f. The ketoacid 4f is not a plausible intermediate in the oxidation of 1f to 3f or to 2f.
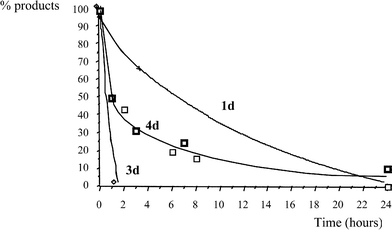 | ||
| Fig. 3 Oxidation of 4-trifluoromethylmandelic acid (1d), 4-trifluoromethylbenzaldehyde (3d) and 4-trifluoromethylphenylglyoxylic acid (4d) catalysed by CoCl2 | ||
| Substrate | Time/h | % Conversion | Products | % Product yield |
|---|---|---|---|---|
| 3d | 2 | 100 | 2d | 60 |
| 4d | 7 | 75 | 2d | 100 |
| 1d | 24 | 100 | 2d | 83 |
| 3d | 17 |
The oxidation of 4-trifluoromethylbenzaldehyde (3d) gave 60% of 4-trifluoromethylbenzoic acid (2d) after complete conversion in 2 h. The oxidation of 4-trifluoromethylphenylglyoxylic acid (4d) afforded exclusively the carboxylic acid 2d with a conversion of 75% after 7 h. In the case of the oxidation of 1d to 2d by CoCl2 occurring with 83% selectivity, both mechanisms, either via path “a” or path “b” in Scheme 2 are compatible with the available data.
We can conclude that the Co-catalysed oxidative decarboxylation of derivatives 1 gives rise to the reaction products 2 and 3 in very different ratios, ranging from 1∶99 to 89∶11, depending on the substrate substitution. The reaction follows different pathways depending on the nature of the aryl group. Thus, for the methoxy-substituted 1f, the aldehyde 3f was selectively obtained following mainly path “b”. For unsubstituted 1a or for CF3-substituted 1d the carboxylic acids 2a and 2d were the main reaction products, respectively, and both pathways “a” and “b” are operative.
Comparative studies between the oxidation of mandelic acid derivatives under molecular oxygen catalysed by CoCl2 and by Bi(0)
The oxidative decarboxylation of substrates 1 has been recently examined in a Bi(0) catalysed reaction in the presence of molecular oxygen in DMSO–AcOH.16 As in the Co catalysed oxidation, compounds 2 and 3 were obtained with selectivities depending on the substitution on the aryl ring. However, on comparing both catalytic systems, we observed that the reactions do not always afford the same main product for the same substituent. In order to better compare the CoCl2 and the Bi(0) systems for the oxidation of 1, Table 4 summarises the results obtained in the Bi(0) catalysed reactions, including conversions, yields of 2 and 3 and selectivities.The comparison of the data of Tables 1 and 4 indicates that from the point of view of the reaction rate and conversion, metallic bismuth is a slightly more efficient catalyst than CoCl2 for the oxidation of 1a, 1f and 1g. Similar reactivities were observed for the oxidation of 4-trifluoromethylmandelic acid (1d). For the remaining cases, CoCl2 showed a better catalytic activity than Bi(0).
However, important changes in the selectivities of both Bi(0) and Co(II) catalysed reactions were observed. When benzaldehyde derivatives 3 are the main compounds formed, as in the case of 1e and 1g, a better selectivity is obtained with CoCl2 as compared to Bi(0). On the contrary, when benzoic acid derivatives 2 are the main products as in the oxidation of 1a, 1c and 1d, better selectivities towards 2 are obtained with Bi(0).
In two cases, the selectivity of the two catalytic systems is completely reversed. Thus, for the oxidation of 4-chloro- and 4-methoxymandelic acid (1b and 1f, respectively), the CoCl2 catalysed oxidation affords mainly the corresponding benzaldehyde derivatives 3b and 3f, whereas in the Bi(0) catalysed oxidation, the corresponding benzoic acid derivatives 2b and 2f are selectively obtained. With the Co(II) system, 4-chlorobenzaldehyde (3b) and 4-anisadehyde (3f) are formed with 71% and 84% selectivities, respectively. With the Bi catalytic system, 4-chloro- and 4-methoxybenzoic acids (2b and 2f) are formed in 98% and 62% yields, respectively.
Scheme 5 illustrates the fact that the oxidation of 1b can be oriented towards the formation of either 2b or 3b, according to the nature of the catalytic system. Thus, 2b or 3b can be selectively obtained by using the Bi(0) or the Co(II) systems, respectively. A similar Scheme 6 can be proposed for the oxidation of 1f, indicating that either the aldehyde 3f or the carboxylic acid 2f can be selectively obtained with CoCl2 or Bi(0) as the catalyst, respectively, though with a lesser selectivity.
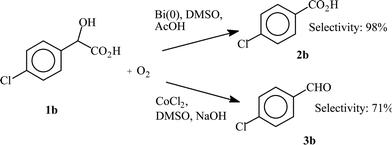 | ||
| Scheme 5 Selectivity in the oxidation of 4-chloromandelic acid (1b) with two different catalytic systems | ||
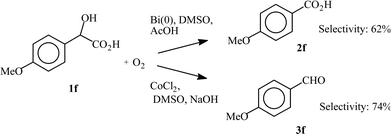 | ||
| Scheme 6 Selectivity in the oxidation of 4-methoxymandelic acid (1f) with two different catalytic systems | ||
For the Bi(0) catalysed oxidations, path “a” via the ketoacid 4 (Scheme 2) was shown to be the main pathway followed for the oxidation of 1a and 1d to the corresponding carboxylic acids 2a and 2d.
Conclusions
The CoCl2/O2 system in DMSO–NaOH constitutes a novel and efficient catalytic system for the oxidative decarboxylation of mandelic acid derivatives. Benzoic acids or benzaldehyde derivatives are obtained selectively, depending on the substituents present on the aromatic ring. In the reactivity examined for a series of para-substituted mandelic acids, it was observed that for OH, OMe and Cl substituents, the oxidation led to selective formation of the corresponding aldehydes 3. In contrast, for H, F and CF3 substituents, the corresponding carboxylic acids 2 were formed in selectivities higher than 80%. The cobalt(II) chloride catalysed oxidation of mandelic acid derivatives is proposed to follow different mechanistic pathways, depending on the substitution on the aryl ring.Interestingly, comparison of the oxidation of the same substrates by a Bi(0)/O2 system presents some similarities, although in the cases of 4-chloromandelic acid and 4-methoxymandelic acid, the main products obtained were different and the selectivities 2∶3 were completely reversed.
Experimental
Commercially available products were used without further purification. HPLC analysis was effectuated with a Waters Millipore apparatus, with a µ-Bondapack C18 Waters 9 µm, 30 cm×3.9 mm column. The eluent was a 80∶20 mixture of H2O–MeOH with H3PO4
(0.5%). The elution was carried out at 1 mL min−1 in isocratic mode and the products were detected by UV at λ=256 nm.
General oxidation procedure
The reactions were carried out under 1 atm molecular oxygen. Mandelic acid or one of its derivatives (2 mmol) was dissolved in DMSO (5 mL) in the presence of anhydrous CoCl2 (0.02 mmol) and NaOH (3 mmol of a 50% aqueous solution). The mixture was stirred at 125°C and the consumption of the starting material was followed by HPLC and/or by 1H NMR. The crude reaction mixture was hydrolysed with 5 mL aqueous 1 M HCl solution saturated with NaCl and extracted with ethyl acetate (5×10 mL). Organic layers were collected and washed twice with an aqueous 0.1 M HCl solution saturated with NaCl, dried over MgSO4 and filtered off. The products were analysed and quantified by HPLC and by 1H NMR and their spectral data compared to those of authentic samples.
References
- J. Iqbal, M. Mukhopadhyay and A. K. Mandal, Synlett, 1997, 876 CAS.
- X. Baucherel, E. Levoirier, J. Uziel and S. Jugé, Tetrahedron Lett., 2000, 41, 1385 CrossRef CAS.
- J. Iqbal and A. Pandey, Tetrahedron Lett., 1990, 31, 575 CrossRef CAS.
- J. Iqbal, M. A. Khan and N. K. Nayyar, Tetrahedron Lett., 1991, 32, 5179 CrossRef CAS.
- D. K. Chand and P. K. Bharadwaj, Inorg. Chem., 1997, 36, 5658 CrossRef CAS.
- A. K. Mandal and J. Iqbal, Tetrahedron, 1997, 53, 7641 CrossRef CAS.
- T. Punniyamurthy, B. Bhatia, M. M. Reddy, G. C. Maikap and J. Iqbal, Tetrahedron, 1997, 53, 7649 CrossRef CAS.
- B. C. Das and J. Iqbal, Tetrahedron Lett., 1997, 38, 1235 CrossRef.
- (a) Y. H. Lin, I. D. Williams and P. Li, Appl. Catal., A, 1997, 150, 221 CrossRef CAS; (b) X. Baucherel, J. Uziel and S. Jugé, J. Org. Chem., 2001, 66, 4504 CrossRef CAS.
- R. S. Drago, B. B. Corden and C. W. Barnes, J. Am. Chem. Soc., 1986, 108, 2453 CrossRef CAS.
- P. Mastrorilli, G. P. Suranna, C. F. Nobile, G. Farinola and L. Lopez, J. Mol. Catal. A: Chem., 2000, 156, 279 CrossRef CAS.
- Y. Yoshino, Y. Hayashi, T. Iwahama, S. Sakaguchi and Y. Ishii, J. Org. Chem., 1997, 62, 6810 CrossRef CAS.
- P. Li and H. Alper, J. Mol. Catal. A, 1992, 72, 143 CrossRef CAS.
- See, for example: (a) C. Walling and K. Amaranath, J. Am. Chem. Soc., 1982, 104, 1185 CrossRef CAS; (b) D. Ip and J. Rocek, J. Am. Chem. Soc., 1979, 101, 6311 CrossRef CAS.
- C. Coin, V. Le Boisselier, I. Favier, M. Postel and E. Duñach, Eur. J. Org. Chem., 2001, 735 CrossRef CAS.
- I. Favier, F. Giulieri, E. Duñach, D. Hébrault and J. R. Desmurs, Eur. J. Org. Chem., 2002, 1984 CrossRef CAS.
- I. Favier and E. Duñach, Tetrahedron, 2003, 59, 1823 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2004 |