Preparation, characterization and electrochemical and X-ray structural studies of new conjugated 1,1′-ferrocenediyl-ended [CpFe-arylhydrazone]+ salts
Carolina
Manzur
*a,
César
Zúñiga
a,
Lorena
Millán
a,
Mauricio
Fuentealba
a,
Jose A.
Mata
b,
Jean-René
Hamon
*c and
David
Carrillo
*a
aLaboratorio de Química Inorgánica, Instituto de Química, Pontificia Universidad Católica de Valparaíso, Avenida Brasil, 2950, Valparaíso, Chile. E-mail: david.carrillo@ucv.cl; Fax: +56 32 27 34 20; Tel: +56 32 27 31 65
bDepartamento de Química Inorgánica y Orgánica, Universitat Jaume I, 12080, Casstellón, Spain
cLaboratoire des Organométalliques et Catalyse: Chimie et Electrochimie Moléculaires CNRS UMR 6509, Institut de Chimie de Rennes, Université de Rennes 1, Campus de Beaulieu, 35042, Rennes cedex, France. E-mail: jean-rene.hamon@univ-rennes1.fr; Fax: +33 223 23 56 37; Tel: +33 223 23 59 58
First published on 14th November 2003
Abstract
A series of new conjugated bimetallic ferrocenyl 1,1′-bis-substituted compounds of the type (E)-[CpFe(η6-p-RC6H4)NHN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(η5-C5H4)Fe(η5-C5H4)–CHCHC6H4-p-R′]+PF6−
(Cp
CH(η5-C5H4)Fe(η5-C5H4)–CHCHC6H4-p-R′]+PF6−
(Cp![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =η5-C5H5; R, R′=H, NO2, 11; Me, NO2, 12; MeO, NO2, 13; Cl, NO2, 14; Me, CN, 15; Me, Me, 16), with end-capped (E)-ethenylaryl and [CpFe(arylhydrazone)]+ substituents, have been prepared by the condensation reaction of 1,1′-(p-R′-arylethenyl)ferrocenecarboxaldehyde (R′=Me, 4; NO2, 5; CN, 6) with the organometallic hydrazine precursors [CpFe(η6-p-RC6H4NHNH2)]+PF6−
(R=H, 7; Me, 8; MeO, 9; Cl, 10). In the trimetallic series, {[CpFe(η6-p-RC6H4)NHNCH(η5-C5H4)]2Fe}2+[PF6−]2
(R=H, 17; Me, 18; MeO, 19, Cl, 20), which results from the condensation of two equivalents of the same organometallic hydrazine precursor (7–10) with 1,1′-ferrocenedicarboxaldehyde, the ferrocenediyl core symmetrically links two cationic mixed-sandwich units. These ten hydrazones (11–20) were stereoselectively obtained as their trans isomers about the NC double bond. All the new compounds were thoroughly characterized by a combination of elemental analysis, spectroscopic techniques (1H NMR, IR and UV-Vis) and electrochemical studies in order to prove electronic interaction between the donating and accepting units through the π-conjugated system. A representative example of each series has also been characterized by single crystal X-ray diffraction analysis. The bimetallic complex 16+ adopts an anti conformation with the two iron atoms on opposite faces of the dinucleating hydrazonato ligand, whereas the trinuclear complex 192+ adopts a syn conformation with an Fe–Fe–Fe angle of 180°. Other salient features of these structures are the long Fe–Cipso bond distances and the slight cyclohexadienyl character at the coordinated C6 ring, with a folding angle of 7.4° and 7.0° for 16+ and 192+, respectively.
=η5-C5H5; R, R′=H, NO2, 11; Me, NO2, 12; MeO, NO2, 13; Cl, NO2, 14; Me, CN, 15; Me, Me, 16), with end-capped (E)-ethenylaryl and [CpFe(arylhydrazone)]+ substituents, have been prepared by the condensation reaction of 1,1′-(p-R′-arylethenyl)ferrocenecarboxaldehyde (R′=Me, 4; NO2, 5; CN, 6) with the organometallic hydrazine precursors [CpFe(η6-p-RC6H4NHNH2)]+PF6−
(R=H, 7; Me, 8; MeO, 9; Cl, 10). In the trimetallic series, {[CpFe(η6-p-RC6H4)NHNCH(η5-C5H4)]2Fe}2+[PF6−]2
(R=H, 17; Me, 18; MeO, 19, Cl, 20), which results from the condensation of two equivalents of the same organometallic hydrazine precursor (7–10) with 1,1′-ferrocenedicarboxaldehyde, the ferrocenediyl core symmetrically links two cationic mixed-sandwich units. These ten hydrazones (11–20) were stereoselectively obtained as their trans isomers about the NC double bond. All the new compounds were thoroughly characterized by a combination of elemental analysis, spectroscopic techniques (1H NMR, IR and UV-Vis) and electrochemical studies in order to prove electronic interaction between the donating and accepting units through the π-conjugated system. A representative example of each series has also been characterized by single crystal X-ray diffraction analysis. The bimetallic complex 16+ adopts an anti conformation with the two iron atoms on opposite faces of the dinucleating hydrazonato ligand, whereas the trinuclear complex 192+ adopts a syn conformation with an Fe–Fe–Fe angle of 180°. Other salient features of these structures are the long Fe–Cipso bond distances and the slight cyclohexadienyl character at the coordinated C6 ring, with a folding angle of 7.4° and 7.0° for 16+ and 192+, respectively.
Introduction
Ferrocenes1 play important roles in the fields of organic and organometallic chemistry and in materials science as components of dendrimers,2 polymers,3 molecular magnets4 and non-linear optical (NLO) materials.5 Their chemistry has been extensively explored because ferrocene-based complexes combine chemical versatility and excellent thermal and photochemical stability with exceptional electrochemical properties, which generate considerable interest in the use of the ferrocenyl moiety, [CpFe(η5-C5H4)] (Cp=η5-C5H5), as a donating group in donor-acceptor (D-π-A) chromophores displaying enhanced second-order NLO properties.5,6 The electron-releasing ability of the parent ferrocenyl group is closely similar to that of the p-methoxyphenyl group.7 On the other hand, the cationic isolobal electron-acceptor counterparts of ferrocene, the robust mixed-sandwich derivatives [CpFe(η6-arene)]+, have also been the subject of intense synthetic,8 electron transfer,9 electrochemical10 and photochemical11 investigations. Despite this interest, only in one case has the [CpFe(η6-aryl)]+ moiety been used as an organometallic chromophore to achieve second-order NLO responses.12 In the search for new materials with electronic communication between terminal subunits, we have focused our interest in the preparation of new conjugated ferrocenyl complexes with end-capped mixed-sandwich iron(II) fragments,13 affording interesting bimetallic complexes in which the organometallic termini are connected by an asymmetric –NR–NCR–
(R=H, Me) type I hydrazonato14 conjugated bridge. Furthermore, non-linear optical responses can be envisaged from electrochemical studies, electronic spectroscopy, molecular structure determinations and theoretical investigations that suggest effective electron delocalization along the π-conjugated spacer.13b,d,e
In a continuation of these studies, we have now prepared conjugated ferrocenyl 1,1′-bis-substituted compounds with end-capped arylethenyl substituents and [CpFe(arylhydrazone)]+ groups for the bimetallic series 11–16 , whereas in the trimetallic series 17–20, the ferrocenediyl core symmetrically links two cationic mixed-sandwich units. The full analytical and spectroscopic characterization (IR, UV-VIS, 1H NMR) of these ten new organometallic hydrazone complexes and of the new 1,1′-(p-tolylethenyl)ferrocenecarboxaldehyde (4) and its protected 1,3-dioxolane precursor 3 (see below) are reported here. Their electronic and electrochemical properties have been investigated and the crystal structures of a representative example of each series, namely compounds 16 and 19, have also been determined. Structurally related dicationic trinuclear organoiron oligomers have recently been prepared by Abd-El-Aziz et al.15 in order to synthesize a new class of mixed-charge iron-containing polymers. In addition, 1,1′- bis-ethenediyl ferrocene complexes with various pendant groups such as pyridine,16p-cyano,17p-bromo18 and p-iodophenylene19 have been used by several authors to synthesize trimetallic derivatives that show very interesting structural features and high non-linear optical responses.
Results and discussion
Synthesis and spectroscopy of 3 and 4
As pointed out by most authors, the E-type conformation of the π-bridge is known to provide a good coplanarity of the conjugated spacer and hence to increase the electronic communication between the donating and accepting ends of a dipolar system.17–21 Therefore, we first attempted to prepare the new bis-substituted ferrocene compound (E)-1,1′-(p-methylstyryl)ferrocene carboxaldehyde (4, Scheme 1) by conventional Wittig methods, the most appropriate strategy for short-length chained complexes, according to the procedure described by Peris and co-workers for the analogous derivatives 5 and 6, bearing the p-nitro and p-cyano electron-withdrawing groups, respectively.17 The reaction afforded a poor yield of an untractable mixture and therefore, in order to obtain our target molecule 4, we applied the multi-step procedure developped by Manoury et al.21 (Scheme 1). This involves the preparation of the monoacetal 2 from 1,1′-ferrocenedicarboxaldehyde 1, which is readily accessible from ferrocene.22 The free aldehyde function was then used to build the first substituent by reacting 2 with the in situ generated (p-MeC6H4CHPPh3) ylide. Surprisingly, compound 3 was stereoselectively formed, in 59% yield, as the E isomer as demonstrated by 1H NMR spectroscopy (see below). The Wittig reaction generally leads to a mixture of E and Z isomers. Hydrolytic deprotection of the 1,3-dioxolane intermediate 3 furnished the desired bis-substituted ferrocene carboxaldehyde 4 in 47% yield (Scheme 1). The new free formyl group has then been used in the building up of the second substituent as described in the following section.
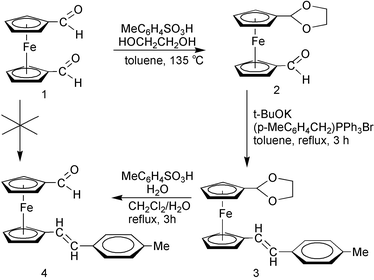 | ||
| Scheme 1 | ||
Characterization of the new products 3 and 4 was mainly achieved by means of IR and 1H NMR spectroscopies and satisfactory elemental analysis (see Experimental). In the proton NMR spectra, both E isomers 3 and 4 are identified by their AB quartet with a coupling constant of ca. 17 Hz, in accord with the expected trans stereochemistry. Four triplets were observed for the substituted Cp rings, while the phenyl protons appear as two doublets. On the other hand, the carboxaldehyde function of compound 4 exhibited the characteristic strong ν(CO) stretching vibration at 1679 cm−1 in the infrared spectrum, and low field singlets at δ 9.91 and δ 192.7 in the 1H and 13C{1H} NMR spectra, respectively.
The cyclovoltammogram of complex 4 displayed the chemically reversible ferrocene/ferricinium couple with ipa/ipc=1.0 in CH2Cl2. The large peak-to-peak separation (196 mV) may be due to a combination of uncompensated solution resistance and slightly slow electron transfer kinetics.23 As expected as the result of substituting an electron-withdrawing group (NO2, CN) for an electron-releasing one (Me), the half-wave potential of the ferrocenediyl core for 4
(267 mV) is less anodic, with respect to ferrocene, than those measured for 5
(310 mV) and 6
(330 mV),17 indicating some degree of electron transfer between the p-Me substituent and the iron center.
Synthesis and spectroscopy of 11–20
The 1-carboxaldehyde-1′-p-R′-(E)-styrylferrocenes (η5-C5H4CHO)Fe(η5-C5H4–(E)–CHCH–C6H4-p-R′)
(R′=Me, 4; NO2, 5; CN, 6) are indeed of interest in the design of new materials, since the presence of the carboxaldehyde group can provide access to a variety of functional groups. Thus, the new dinuclear organoiron hydrazones 11–16
(see Experimental and Table 1) were successfully prepared by a condensation reaction of the ionic organometallic hydrazine precursors [CpFe(η6-p-RC6H4NHNH2)]+PF6−
(R=H, 7; Me, 8; MeO, 9; Cl, 10) with the ferrocene-based aldehyde building blocks 4–6
(Scheme 2).
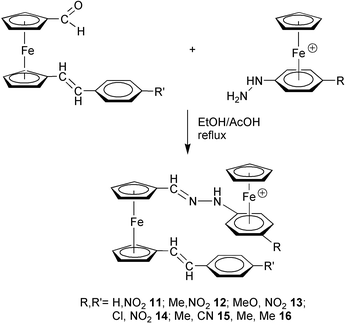 | ||
| Scheme 2 | ||
| Compound | R, R′ | [CpFe(p-RC6H4NHNH2)]+PF6− | (C5H4CHO)Fe(C5H4–CHCHC6H4-p-R′ |
Yield |
|---|---|---|---|---|
| a The reactions listed in this table were all run under the general conditions given in the Experimental. | ||||
| 11 | H, NO2 | 49.8 mg, 0.138 mmol | 49.8 mg, 0.138 mmol | 55%, 54 mg |
| 12 | Me, NO2 | 53.6 mg, 0.138 mmol | 49.8 mg, 0.138 mmol | 58%, 58 mg |
| 13 | MeO, NO2 | 55.8 mg, 0.138 mmol | 49.8 mg, 0.138 mmol | 55%, 57 mg |
| 14 | Cl, NO2 | 56.5 mg, 0.138 mmol | 49.8 mg, 0.138 mmol | 53%, 55 mg |
| 15 | Me, CN | 100.0 mg, 0.258 mmol | 87.0 mg, 0.255 mmol | 40%, 72 mg |
| 16 | Me, Me | 50.0 mg, 0.129 mmol | 43.0 mg, 0.130 mmol | 56%, 50 mg |
On the other hand, the trimetallic hydrazones [{CpFe(η6-p-RC6H4NHNCH–η5-C5H4)}2Fe]2+[PF6−]2
(17–20; see Experimental and Table 2) were synthesized by the reaction of 2 equiv. of organoiron hydrazine precursors 7–10 with 1,1′-ferrocenedicarboxaldehyde (η5-C5H4CHO)2Fe (Scheme 3). It is interesting to note that the bis functionalized p-chloro derivative 20 represents an attractive monomeric unit for the preparation of polymers containing both neutral and cationic cyclopentadienyliron complexes in their structures, in light of the recently communicated synthetic methodology.15
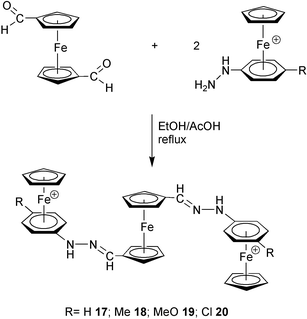 | ||
| Scheme 3 | ||
| Compound | R | [CpFe(p-RC6H4NHNH2)]+PF6− | (C5H4CHO)2Fe | Yield |
|---|---|---|---|---|
| a The reactions listed in this table were all run under the general conditions given in the Experimental. | ||||
| 17 | H | 101.0 mg, 0.270 mmol | 32.0 mg, 0.132 mmol | 55%, 69 mg |
| 18 | Me | 109.5 mg, 0.282 mmol | 34.0 mg, 0.140 mmol | 51%, 70 mg |
| 19 | MeO | 115.1 mg, 0.285 mmol | 34.6 mg, 0.143 mmol | 50%, 73 mg |
| 20 | Cl | 51.5 mg, 0.126 mmol | 15.0 mg, 0.062 mmol | 59%, 37 mg |
In all cases, the reactions were carried out in refluxing ethanol solution containing catalytic amounts of concentrated acetic acid. The bi- and trinuclear complexes were obtained as spectroscopically pure air- and thermally stable, brownish red to dark red (11–16) and orange or red-orange (17–20) microcrystalline solids in reasonable yields ranging from 40 to 59%, after recrystallization from CH2Cl2–Et2O (1∶1). The cationic compounds exhibit a good solubility in common polar organic solvents, but are insoluble in diethyl ether, hydrocarbons and water. The structures of these ten new homobi- and homotrimetallic hydrazones were inferred from satisfactory elemental analyses (Table 3), 1H NMR, IR and UV-Vis spectroscopies (Table 4) and, additionally, in the case of complexes 16 and 19, by X-ray diffraction analysis (vide infra).
| Compound | Color | M. p.a/°C | Analyses |
|---|---|---|---|
| a All the compounds decompose when melted. | |||
| 11 | Brownish red | 149 | Found: C, 49.88; H, 3.87; N, 5.76 |
| Calcd.: C, 50.24; H, 3.65; N, 5.86 | |||
| 12 | Brownish red | 105 | Found: C, 51.12; H, 4.00; N, 5.67 |
| Calcd.: C, 50.92; H, 3.86; N, 5.75 | |||
| 13 | Brownish red | 97 | Found: C, 50.29; H, 3.89; N, 5.94 |
| Calcd.: C, 49.83; H, 3.78; N, 5.62 | |||
| 14 | Brownish red | 187 | Found: C, 48.24; H, 3.56; N, 5.98 |
| Calcd.: C, 47.94; H, 3.35; N, 5.59 | |||
| 15 | Dark red | 155 | Found: C, 53.79; H, 4.12; N, 6.33 |
| Calcd.: C, 54.04; H, 3.97; N, 5.91 | |||
| 16 | Dark red | 215 | Found: C, 55.25; H, 4.61; N, 4.09 |
| Calcd.: C, 54.89; H, 4.46; N, 4.00 | |||
| 17 | Orange | 209 | Found: C, 43.13; H, 3.40; N, 6.06 |
| Calcd.: C, 42.80; H, 3.38; N, 5.87 | |||
| 18 | Orange | 263 | Found: C,43.78; H, 3.78; N, 5.92 |
| Calcd.: C, 44.02; H, 3.69; N, 5.70 | |||
| 19 | Orange | 240 | Found: C, 42.80; H, 3.64; N, 5.79 |
| Calcd.: C, 42.64; H, 3.58; N, 5.52 | |||
| 20 | Red-orange | 215 | Found: C, 40.32; H, 3.17; N, 5.39 |
| Calcd.: C, 39.92; H, 2.96; N, 5.48 | |||
| Compound | IR (KBr) ν/cm−1 | UV-vis λ/nm (Log ε/dm3 mol−1 cm−1) | 1H NMRaδ (CD3COCD3) |
|---|---|---|---|
| a Recorded at 200 MHz for 11–16 and at 400 MHz for 17–20. | |||
| 11 | 3328 m (NH), 1590 m (CC), 1570 m (CN), 1333 s (NO2), 840 vs and 558 m (PF6) |
CH2Cl2: 322 (4.40), 489 (3.71) | 4.55 (br s, 4H, C5H4), 4.81 (br s, 2H, C5H4), 4.86 (br s, 2H, C5H4), 5.03 (s, 5H, Cp), 6.10–6.21 (m, 5H, coord Ph), 6.88 (d, JH-H=16.3, 1H, CHCH), 7.16 (d, JH-H=16.2, 1H, CHCH), 7.60 (d, JH-H=8.8, 2H, Ph), 7.88 (s, 1H, NCH), 8.00 (d, JH-H=8.9, 2H, Ph), 9.42 (br s, 1H, NH) |
| DMSO: 332 (4.33), 488 (3.62) | |||
| 12 | 3328 m (NH), 1588 m (CC), 1570 m (CN), 1338 s (NO2), 840 vs and 557 m (PF6) |
CH2Cl2: 321 (4.36), 351sh (4.32), 492 (3.67) | 2.46 (s, 3H, CH3), 4.53 (br s, 4H, C5H4), 4.80 (t, JH-H=1.7, 2H, C5H4), 4.83 (t, JH-H=1.7, 2H, C5H4), 4.96 (s, 5H, Cp), 6.05–6.08 (m, 4H, coord Ph), 6.87 (d, JH-H=16.2, 1H, CHCH), 7.15 (d, JH-H=16.2, 1H, CHCH), 7.60 (d, JH-H=8.8, 2H, Ph), 7.88 (s, 1H, NCH), 7.98 (d, JH-H=8.9, 2H, Ph), 9.43 (br s, 1H, NH) |
| DMSO: 327 (4.33), 341sh (4.32), 501 (3.62) | |||
| 13 | 3330 m (NH), 1590 m (CC), 1570 m (CN), 1337 s (NO2), 1256 s (C–O), 840 vs and 557 m (PF6) |
CH2Cl2: 315 (4.31), 357sh (4.25), 487 (3.64) | 4.02 (s, 3H, CH3O), 4.53 (br s, 4H, C5H4), 4.80 (t, JH-H=1.7, 2H, C5H4), 4.83 (t, JH-H=1.7, 2H, C5H4), 5.00 (s, 5H, Cp), 6.00 (d, JH-H=6.9, 2H, coord Ph), 6.12 (d, JH-H=6.9, 2H, coord Ph), 6.88 (d, JH-H=16.2, 1H, CHCH), 7.15 (d, JH-H=16.1, 1H, CHCH), 7.61 (d, JH-H=8.7, 2H, Ph), 7.86 (s, 1H, NCH), 8.01 (d, JH-H=8.7, 2H, Ph), 9.36 (br s, 1H, NH) |
| DMSO: 332 (4.35), 351 (4.32), 490 (3.69) | |||
| 14 | 3324 m (NH), 1590 m (CC), 1558 m (CN), 1340 s (NO2), 1098 w (C–Cl), 836 vs and 556 s (PF6) |
CH2Cl2: 323 (4.44), 355 (4.39), 487 (3.79) | 4.53 (t, JH-H=1.7, 2H, C5H4), 4.56 (t, JH-H=1.7, 2H, C5H4), 4.81 (t, JH-H=1.7, 2H, C5H4), 4.85 (t, JH-H=1.7, 2H, C5H4), 5.12 (s, 5H, Cp), 6.18 (d, JH-H=6.8, 2H, coord Ph), 6.53 (d, JH-H=6.6, 2H, coord Ph), 6.87 (d, JH-H=16.2, 1H, CHCH), 7.14 (d, JH-H=16.3, 1H, CHCH), 7.60 (d, JH-H=8.7, 2H, Ph), 7.89 (s, 1H, NCH), 8.01 (d, JH-H=8.8, 2H, Ph), 9.59 (br s, 1H, NH) |
| DMSO: 331 (4.38), 462 (3.71) | |||
| 15 | 3331 w (NH), 2221 m (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1620 w (CC), 1572 m (CN), 838 vs and 557 s (PF6) N), 1620 w (CC), 1572 m (CN), 838 vs and 557 s (PF6) |
CH2Cl2: 250 (4.26), 286 (3.96), 311 (4.04), 343 (4.12), 407 (3.33), 478 (3.27) | 2.47 (s, 3H, CH3), 4.49 (m, 4H, C5H4), 4.78 (m, 4H, C5H4), 4.91 (s, 5H, Cp), 6.00 (d, JH-H=5.2, 2H, coord Ph), 6.09 (d, JH-H=5.3, 2H, coord Ph), 6.74 (d, JH-H=16.4, 1H, CHCH), 7.00 (d, JH-H=16.2, 1H, CHCH), 7.46 (s, 4H, Ph), 7.80 (s, 1H, NCH), 9.30 (br s, 1H, NH) |
| DMSO: 316 (4.24), 353 (4.11), 428 (3.17), 479 (3.26) | |||
| 16 | 3331 w (NH), 1620 w (CC), 1572 m (CN), 838 vs and 557 s (PF6) |
CH2Cl2: 274 (4.03), 302 (3.15), 327 (3.45), 360 (3.08), 453 (3.04) | 2.34 (s, 3H, CH3), 2.52 (s, 3H, CH3), 4.55 (br s, 4H, C5H4), 4.84 (br s, 4H, C5H4), 4.99 (s, 5H, Cp), 6.08 (d, JH-H=6.2, 2H, coord Ph), 6.17 (d, JH-H=6.3, 2H, coord Ph), 6.60 (d, JH-H=16.0, 1H, CHCH), 6.77 (d, JH-H=16.1, 1H, CHCH), 7.02 (d, JH-H=7.9, 2H, Ph), 7.23 (d, JH-H=7.8, 2H, Ph), 7.86 (s, 1H, NCH), 9.26 (br s, 1H, NH) |
| DMSO: 261 (4.38), 308 (3.93), 331 (4.32), 418 (3.13), 465 (3.49) | |||
| 17 | 3316 w (NH), 1570 s and 1560 s (CN), 842 vs, 831 vs and 558 m (PF6) |
CH2Cl2: 240(3.95), 277(3.30), 312(3.85), 351(3.45), 410(3.09), 491 (2.92) | 4.54 (br s, 4H, C5H4), 4.85 (br s, 4H, C5H4), 5.01 (s, 10H, Cp), 6.06–6.51 (m, 10H, Ph), 8.00 (s, 2H, NCH), 9.54 (s, 2H, NH) |
| DMSO: 319 (3.84), 356 (3.59), 419 (3.15), 466 (3.05) | |||
| 18 | 3336 m (NH), 1568 s (CN), 844 vs, 826 vs and 558 s (PF6) |
CH2Cl2: 239 (4.15), 274 (3.40), 309 (3.98), 349 (3.71), 407 (3.16), 473 (3.13) | 2.44 (s, 6H,CH3), 4.54 (br s, 4H, C5H4), 4.85 (br s, 4H, C5H4), 4.94 (s, 10H,Cp), 6.10 (br s, 8H, Ph), 7.83 (s, 2H, NCH), 9.23 (s, 2H, NH) |
| DMSO: 272 (3.47), 315 (3.46), 355 (3.34), 407 (2.62), 457 (2.76) | |||
| 19 | 3344 m (NH), 1570 s (CN), 1252 s (C–O), 841 vs and 558 s (PF6) |
CH2Cl2: 242 (3.88), 274 (3.78), 313 (3.80), 354 (3.61), 404 (3.00), 470 (2.99) | 3.98 (s, 6H, CH3O), 4.53 (br s, 4H, C5H4), 4.83 (br s, 4H, C5H4), 4.98 (s, 10H,Cp), 6.03 (d, JH-H=6.9, 4H, Ph), 6.11 (d, JH-H=7.1, 4H, Ph), 7.79 (s, 2H, NCH), 9.17 (s, 2H, NH) |
| DMSO: 270 (3.80), 319 (3.61), 360 (3.46), 417 (3.16), 468 (3.01) | |||
| 20 | 3329 m (NH), 1570 s and 1560 s (CN), 1092 w and 1062 w (C–Cl), 843 vs, 829 vs and 557 s (PF6) |
CH2Cl2: 240 (3.86), 290 (2.88), 302 (3.59), 345 (3.56), 416 (2.91), 487 (2.89) | 4.54 (br s, 4H, C5H4), 4.85 (br s, 4H, C5H4), 5.10 (s, 10H, Cp), 6.23 (br s, 4H, Ph), 6.53 (br s, 4H, Ph), 7.83 (s, 2H, NCH), 9.49 (s, 2H, NH) |
| DMSO: 274 (3.81), 324 (3.79), 359 (3.43), 433 (3.13) | |||
The most remarkable common features observed in the IR spectra of these ten powdered compounds (11–20 in Table 4) were: (i) the existence of a sharp intense band at ca. 1570 cm−1, which is due to the asymmetric stretching vibration of the CN imine group, (ii) the typical weak to medium N–H stretching vibration in the region from 3316 to 3344 cm−1 and (iii) a very strong ν(PF6) band at ca. 840 cm−1 and a sharp and strong δ(P–F) band at 557–559 cm−1. In addition, the infrared spectra of compounds 11–16 showed a weak band in the 1588–1620 cm−1 region assigned to the ν(CC) stretching mode of the phenylethenyl pendant group. More specifically, the IR spectra of compounds 11–14 exhibited a strong band in the range 1333–1340 cm−1 attributed to the p-NO2 substituent, whereas the CN stretching vibration appeared at 2221 cm−1 in the IR spectrum of 15. Finally, the spectra of the homotrimetallic compounds 17 and 20 showed two NC, PF6− (and C–Cl for 20) stretching frequencies (see Table 4), a phenomenon that we have already encountered for homobimetallic ferrocenyl hydrazone complexes, which is compatible with the presence of at least two conformers in the solid state.13b
Interestingly, the organoiron hydrazones 11–20 are stereoselectively formed as the sterically less hindered trans (about the NC double bond) isomer as indicated by the unique set of signals in their 1H NMR spectra (acetone-d6, 297 K, see Table 4) and definitively assigned from the structural analyses of 16 and 19
(see below). For all the compounds, the two sandwich moieties [CpFe(η6-p-RC6H4–)]+ and [(η5-C5H4)2Fe] are clearly identified by the characteristic sharp singlet of the Cp proton resonance observed at ca. 5.0 ppm while the two monosubstituted C5 rings appeared as broad singlets or unresolved triplets in the 4.80–4.86 and 4.53–4.56 ppm ranges, corresponding to the spectrum of an A2B2 system. Only one pair of Hα and Hβ resonances is observed for the trimetallic compounds 17–20, consistent with a symmetrical structure with an inversion center at the ferrocenediyl iron atom, in agreement with the solid state structure of 19
(see below). The p-substituted phenylethenyl pendant groups of the bimetallic derivatives 11–16 exhibited the same resonance and coupling constant patterns as those reported above for the ferrocene-based aldehyde 4, indicating that the trans stereochemistry about the HCCH double bond is retained after the construction of the hydrazone framework. The low field position (δ 9.26–9.59) of the acidic benzylic N–H24 signal may be attributed to the electronic effect of the organometallic moiety,8a,25 and/or its participation in molecular association via intermolecular H-bonding.13d Finally, the upfield-shifted aromatic protons of the coordinated C6 ring and the deshielded sharp proton resonance at ca. 7.90 ppm assigned to the azomethine fragment (NCH) appeared in the expected regions.13
Crystal structures
The molecular structures of the cationic (E)-[CpFe(η6-p-MeC6H4)–NHNCH–(η5-C5H4)Fe(η5-C5H4)–CHCH–C6H4-p-Me]+
(16+) and of the dicationic [{CpFe(η6-p-MeOC6H4)–NHNCH–(η5-C5H4)}2Fe]2+
(192+) organometallic moieties, along with the atom labelling schemes, are presented in Fig. 1 and Fig. 2, respectively. Key bond lengths and angles for 16+ and 192+ are listed in Table 5. In both complexes, high anisotropic thermal motion and/or disorder was observed for the carbon atoms of the Cp ligand of the mixed-sandwich units. Such a phenomenon has frequently been reported for mixed-sandwich iron(II) complexes.13b, 13c, 24, 26, 27 The orientation is presumably due to partial rotation of the C5 ring about the Fe–Cp centroid axis. The metrical parameters (see Table 5) are typical of η5–Fe–η6 and η5–Fe–η5 metallocene-type coordination.28–31 The carbocyclic rings coordinated to the same iron center are essentially parallel with one another and the ring centroid–iron–ring centroid vectors are almost collinear, both in the [Cp–Fe–arene]+ and in the ferrocenic subunits.
![Molecular structure and atom numbering scheme for the cation (E)-[CpFe(η6-p-MeC6H4)–NHNCH–(η5-C5H4)Fe(η5-C5H4)–CHCH–C6H4-p-Me]+
(16+). Hydrogen atoms and counter anion PF6− have been omitted for clarity. Displacement ellipsoids are at the 50% probability level.](/image/article/2004/NJ/b308626g/b308626g-f1.gif) | ||
| Fig. 1 Molecular structure and atom numbering scheme for the cation (E)-[CpFe(η6-p-MeC6H4)–NHNCH–(η5-C5H4)Fe(η5-C5H4)–CHCH–C6H4-p-Me]+
(16+). Hydrogen atoms and counter anion PF6− have been omitted for clarity. Displacement ellipsoids are at the 50% probability level. | ||
![Molecular structure and atom numbering scheme for the dication [{CpFe(η6-p-MeOC6H4)–NHNCH–(η5-C5H4)}2Fe]2+
(192+). Hydrogen atoms and counter anion PF6− have been omitted for clarity. Displacement ellipsoids are at the 50% probability level.](/image/article/2004/NJ/b308626g/b308626g-f2.gif) | ||
| Fig. 2 Molecular structure and atom numbering scheme for the dication [{CpFe(η6-p-MeOC6H4)–NHNCH–(η5-C5H4)}2Fe]2+
(192+). Hydrogen atoms and counter anion PF6− have been omitted for clarity. Displacement ellipsoids are at the 50% probability level. | ||
| 16+ | 19+ | 16 | 19 | ||
|---|---|---|---|---|---|
|
a Abbreviations: Cp=η5-C5H5, Cp′=η5-C5H4, Ph=η6-C6H4, CNT=centroid.
b Average distance.
c Sum of the bond distances.
d Measured through-space distance.
|
|||||
| C–C(1–5)b | 1.308 | 1.346 | C–C(6–11)b | 1.406 | 1.392 |
| C(9)–O(1) | – | 1.360(5) | C(6)–N(1) | 1.359(6) | 1.366(4) |
| N(1)–N(2) | 1.383(5) | 1.382(4) | N(2)–C(13) | 1.278(6) | 1.275(4) |
| C(13)–C(14) | 1.448(7) | 1.421(5) | C–C(14–18)b | 1.415 | 1.411 |
| C–C(19–23)b | 1.412 | – | C(19)–C(24) | 1.455(8) | – |
| C(24)–C(25) | 1.296(7) | – | C(25)–C(26) | 1.501(8) | – |
| Fe(1)–C(1–5)b | 2.011 | 2.045 | Fe(1)–C(6) | 2.193(5) | 2.144(4) |
| Fe(1)–C(7) | 2.087(5) | 2.073(4) | Fe(1)–C(8) | 2.064(5) | 2.048(4) |
| Fe(1)–C(9) | 2.084(5) | 2.116(5) | Fe(1)–C(10) | 2.056(5) | 2.077(4) |
| Fe(1)–C(11) | 2.098(5) | 2.064(4) | Fe(2)–C(14–18)b | 2.050 | 2.039 |
| Fe(2)–C(19–23)b | 2.048 | – | Fe(1)⋯Fe(2)c | 9.714 | 9.633 |
| Fe(1)⋯Fe(2)d | 8.064 | 6.925 | Fe(1)–CpCNT | 1.675 | 1.658 |
| Fe(1)–PhCNT | 1.565 | 1.556 | Fe(2)–CpCNT | 1.658 | 1.649 |
| Fe(2)–Cp′CNT | 1.659 | – | |||
| C(6)–N(1)–N(2) | 120.7(4) | 120.5(4) | N(1)–N(2)–C(13) | 114.9(4) | 117.2(4) |
| N(2)–C(13)–C(14) | 121.2(5) | 123.2(5) | C(19)–C(24)–C(25) | 127.5(6) | – |
| C(24)–C(25)–C(26) | 126.5(6) | – | CpCNT–Fe(1)–PhCNT | 179.0 | 179.2 |
| Cp′CNT–Fe(2)–CpCNT | 178.9 | – | |||
Complex 16+ adopts an anti conformation13b with the two iron atoms on opposite faces of the dinucleating hydrazonato ligand (Fig. 1), whereas the trinuclear complex 192+ adopts a syn conformation13b with an Fe(1)–Fe(2)–Fe(1a) angle of 180°
(Fig. 2). Both [CpFe(arene)]+ moieties occupy opposite sites and they are related by symmetry through the inversion center of the molecule. In both complexes, the π-conjugated bridging ligands, [p-RC6H4–NHNCH–C5H4]−
(R=Me, 16+; MeO, 192+), are similar and, therefore, the through-bond Fe⋯Fe distances (9.714 and 9.633 Å, respectively) are closely related, whereas the through-space Fe⋯Fe distance is much longer in 16+
(8.064 Å) than in 192+
(6.925 Å). In complex 16+ the ferrocenylidene groups adopt an eclipsed conformation whereas the staggered one is observed for complex 192+
(see Figs. 1 and 2). The conformation adopted by the dinuclear complex 16+ allows a certain interaction between the coordinated and free phenyl groups, although the dihedral angle between them is 41.65°
(see Fig. 1). On the other hand, the coordinated and non-coordinated phenyl groups are not coplanar with the corresponding Cp and Cp′ ligands of the ferrocenylidene group, subtending dihedral angles of 21.31° and 18.96°, respectively. The phenyl group of the trimetallic species is not coplanar with the Cp ligand, subtending a dihedral angle of 27.70°
(see Fig. 2). These significant but not large deviations from planarity might, however, allow an efficient π-electron delocalization or electronic interaction between the electron-donating and electron-accepting termini through the entire hydrazonato skeleton.
A careful examination of Table 5 reveals some interesting structural features provoked by the strong electron-withdrawing effect of CpFe+ on the arene ligand in both complexes. Thus, the Fe(1)–C(6) bond lengths, 2.193(5) and 2.144(4) Å for 16+ and 192+, respectively, are longer than the mean value of the other Fe(1)–C(C6 ring) bond lengths (see Table 5). Such an Fe–C elongation has already been reported by us for the binuclear organometallic hydrazones.13b,13c This remarkable elongation is a consequence of a partial delocalization of the electron pair located on the N(1) atom toward the mixed-sandwich moiety and is reflected by (i) a depyramidalization of the N(1) atom with typical bond angles of an sp2-hybridized nitrogen atom (Table 5), (ii) a C(6)–N(1) bond length of 1.359(6) Å for 16+ and 1.366(4) Å for 192+, values that are intermediate between those of a single and a double carbon–nitrogen bond31 and (iii) a slight cyclohexadienyl-like character at the coordinated phenyl ring with a folding angle of 7.4° and 7.0° about the C(7)–C(11) axis for both complexes, respectively. Likewise, the Fe(1)–C(9) bond length of 2.116(5) Å in complex 192+ can be also attributed to a partial delocalization of one oxygen lone pair toward the arene ligand, provoking a folding angle around the C(8)–C(10) axis of 5.8°. Hence, the arene ligand adopt a slightly bowed conformation, with the C(6) and C(9) atoms being bent out of the carbocyclic diene plane.
Electronic spectra
Electronic absorption spectra for the cationic bi- and trinuclear hydrazones 11–20 were measured in CH2Cl2 and DMSO; the values are recorded in Table 4. In each solvent the complexes exhibited similar spectra, revealing their isostructural features with two sets of absorption maxima typical of conjugated ferrocenyl systems. The lower energy band (400–500 nm) is assigned to a metal-to-ligand charge-transfer transition (MLCT) and the higher lying absorption in the range of 300 to 360 nm as an intraligand π-π* transition (ILCT). In fact, both bands have some d-d character.32–34 These assignments are in agreement with previously reported theoretical35 and experimental data.17,19–21,36,37 The energy and intensity of these transitions are influenced by the nature of the ancillary ligands, giving rise to bathochromic shifts in its absorption maximum as the result of a lowering of the energy of the π* orbital of the ligand.5a,37a The effect can be further amplified by increasing the acceptor strength of the electron deficient center. Examination of the UV-vis data (Table 4) for the series of four compounds containing the same [(η5-C5H4)Fe(η5-C5H4)–CHN–NH–(η6-p-MeC6H4)FeCp]+PF6− framework and bearing various pendant accepting groups, such as (p-CHCH–C6H4NO2) (12), (p-CHCH–C6H4CN) (15), (p-CHCH–C6H4Me) (16) and [CpFe+(η6-p-MeC6H4NHNCH)] (18), indicates that the energies and the oscillator strengths of both the low and high energy bands are sensitive to the acceptor strength. Thus, while compound 16 exhibits λmax at 302 and 453 nm, these values progressively shift to longer wavelengths in going to 18
(309 and 473 nm), 15
(311 and 478 nm) and finally to 12
(321 and 492 nm). Similar trends are observed for other families of ferrocenyl p-cyanophenylethenyl and phenylethynyl compounds,36c,38 as well as for cationic organoiron polymers containing azo-dye-functionalized side chains.39
The two characteristic CT bands of the dinuclear hydrazones 11–13, 15 and 16 exhibit bathochromic shifts (Fig. 3) on changing from CH2Cl2
(ε=8.90) to DMSO (ε=47.6), indicating increased polarity in the excited state. The more intense π-π* band is more solvatochromic (4–22 nm) than the lower energy band. For the dinuclear hydrazone 14 and the trinuclear compounds 17–20, the higher energy band is also red-shifted (6–22 nm) but the lower energy band shows a negative solvatochromism (−2 to −55 nm, see Fig. 3). This results from a better stabilization of the equilibrium ground state than the Franck–Condon excited state, due to solvation by solvents of increasing polarity.40 Such a negative solvatochromism (hypsochromic shift) is mainly observed for cationic derivatives,13b,13d,37 since in these species the ground state dipole moments are opposite in sign to those of the neutral species,41 due to the acceptor-localized positive charges, but the charge transfer is in the same direction as in the neutral species.
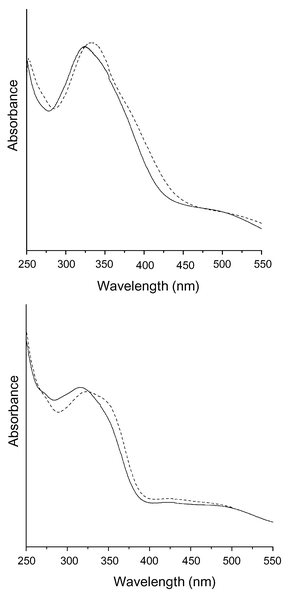 | ||
| Fig. 3 Electronic spectra of 12 (left) and 20 (right), recorded in CH2Cl2 (full line) and in DMSO (dashed line). | ||
It is worth noting that the lower energy maxima for the dicationic ferrocenediyl hydrazones 17–20 are found to be somewhat blue-shifted compared to those of the neutral bis-substituted ferrocenediyl-ligated phenylethenyl derivatives, Fe{(η5-C5H4)-(E)-CHCHC6H4-p-R}2
(R=NO2, CN), reported by Peris et al.17 Coordination of the arene rings with two [CpFe+] fragments is expected to generate stronger dicationic electron-accepting termini. The half-wave oxidation potentials of trimetallic hydrazone derivatives are indeed slightly more anodic than those of the mononuclear neutral species. A rather similar situation has been pointed out by Heck and co-workers37b and two factors were argued to explain this apparent inconsistency: (i) the generation of new orbital frontiers, with changes in character and order, upon complexation and (ii) the molecular orbitals involved in the oxidation step and in the photochemical excitation can be different.
Electrochemical studies
In order to get a deeper insight into the mutual donor-acceptor electronic influence with respect to electrochemical perturbations, we have undertaken the cyclic voltammetry (CV) study of the homobi- and homotrimetallic hydrazone complexes (11–16 and 17–20, respectively). All the cyclovoltammograms were recorded in DMF using the same setup and the electrochemical data thus obtained are summarized in Table 6. All the complexes display two major features: (i) an irreversible process attributable to Fe-centered reduction at the mixed-sandwich fragment10,33b and (ii) a single chemically reversible oxidation wave corresponding to the ferrocenyl unit42 in the bridging linker (Fig. 4). As for the ferrocenediyl-based aldehyde precursor 4, the peak-to-peak separations (ΔEp, see Table 6) are significantly greater than the ideal value of 59 mV required for a pure Nernstian process, probably due to the same reasons cited above.23 However, this difference is similar to that measured for ferrocene under the conditions of the experiment (see Experimental for details).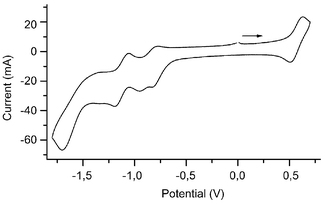 | ||
| Fig. 4 Cyclic voltammograms of complex 12 recorded in DMF+0.1 M n-Bu4N+PF6− at T=293 K and with a voltage sweep rate v=0.1 V s−1, reference electrode Ag/AgCl, internal reference Cp2Fe0/+. | ||
| Compound | E ½[(C5H4)2Fe]/V (ΔEp/mV) | E pc[CpFe+(aryl)]b/V | E ½(p-C6H4NO2)c/V (ΔEp/mV) |
|---|---|---|---|
| a Recorded in DMF at 298 K with a vitreous carbon working electrode, 0.1 M n-Bu4+PF6− as supporting electrolyte, scan rate 100 mV s−1. All potentials are quoted in V vs. the Cp2Fe0/+ couple used as internal reference. b Irreversible wave corresponding to the Fe(II)/Fe(I) couple. c Reversible waves corresponding to the reduction of the nitrophenyl pendant group. | |||
| 11 | 0.176 (98) | −2.25 | −1.36 (131) |
| −1.53 (70) | |||
| 12 | 0.161 (92) | −2.31 | −1.32 (72) |
| −1.53 (70) | |||
| 13 | 0.167 (90) | −2.30 | −1.30 (100) |
| −1.53 (60) | |||
| 14 | 0.197 (109) | −2.31 | −1.31 (72) |
| −1.51 (70) | |||
| 15 | 0.221 (93) | −2.27 | – |
| 16 | 0.045 (102) | −2.53 | – |
| 17 | 0.093 (71) | −2.59 | – |
| 18 | 0.077 (84) | −2.52 | – |
| 19 | 0.190 (83) | −2.48 | – |
| 20 | 0.225 (90) | −2.30 | – |
| −2.44 | |||
By comparison with the previously reported monocationic series, [CpFe(η6-p-RC6H4)NHNCH(η5-C5H4)FeCp]+,13b,43 the bis-substituted complexes show a greater anodic shift (ca. 20–100 mV) of their half-wave potentials, except for 16 as expected with its donating p-Me group and, more surprisingly, 18
(see Table 6). This suggests that the extra electron-accepting substituent has a significant electronic effect on the ferrocenyl bridging spacer. Also surprising is the more anodic half-wave potential of the nitrile derivative 15 compared to that of the corresponding nitro counterpart 12, despite the known better electron-accepting nature of the nitro group. This was already noted for the precursor aldehydes 5 and 6.17 The substitution of the arylethenyl groups by the corresponding organometallic hydrazone ligands, 13vs.19, 14vs.20, 16vs.18, results in a shift of the oxidation wave to more anodic potential, presumably due to the powerful electron-accepting nature of the cationic mixed-sandwich unit.
On the reduction side, the ten compounds studied undergo an irreversible process (Fig. 4) centered at the mixed-sandwich moiety, corresponding to the single-electron reduction of the d6, Fe(II), 18-electron complexes to the unstable d7, Fe(I), 19-electron species.10,33b The very cathodic potential values (Table 6) are very similar to those reported for the monometallic benzaldehyde hydrazone24 and homobimetallic elongated hydrazone-linker-containing complexes13e and are presumably due to the reduction of an in situ generated neutral zwitterionic species.24,44 As expected for the symmetric homotrimetallic compounds 17–20, the integrated area of the reduction wave is twice that of the oxidation one. Note that the cyclovoltammogram of 20 exhibits two reduction waves at −2.30 and −2.44 V vs. Cp2Fe0/+, the more anodic one being attributable to a dechlorination reaction.45,46
On the other hand, the cyclovoltammograms of the four appended p-NO2 substituent derivatives 11–14 exhibit two fully reversible reduction waves at ca. −1.32 and −1.53 V vs. Cp2Fe0/+. The first wave is confidently assigned to the monoelectronic reduction of the nitro group to generate the radical anion –NO2˙−,47,48 whereas the assignment of the second reversible wave remains much more puzzling.49 Nevertheless, this indicates that the LUMO for complexes 11–14 is localized at the nitro substituent. For compounds 15–20, however, the LUMO is determined by the cationic mixed-sandwich, and in all the cases, the nature of the HOMO is dominated by the neutral donating ferrocenyl units, in accordance with theoretical investigations.13b All these electrochemical data taken together suggest that the electron-donating and -accepting termini communicate electronically to a significant extent and that they behave as a donor-acceptor couple.
Concluding remarks
In conclusion, we have described the easy access to two new homogeneous series of conjugated ferrocenyl 1,1′-bis-substituted compounds with end-capped arylethenyl substituents and [CpFe(arylhydrazone)]+ groups for the bimetallic series 11–16 whereas in the trimetallic series 17–20, the ferrocenediyl core symmetrically links two cationic mixed-sandwich units, via condensation reactions between cyclopentadienyliron-complexed hydrazines and ferrocene mono- and biscarbaldehydes, respectively. Single crystal X-ray diffraction analyses show that the bimetallic complex 16+ adopts an anti conformation with the two iron atoms on opposite faces of the dinucleating hydrazonato ligand, whereas the trinuclear complex 192+ adopts a syn conformation with a linear Fe–Fe–Fe arrangement. The ten new organometallic hydrazones 11–20 can be defined as type I non-rod-shaped dipolar chromophores.14 The cationic mixed-sandwich [CpFe(η6-p-RC6H4)–]+ acts as an electron acceptor and the neutral ferrocenediyl moiety as an electron-donating group. Their mutual electronic influence is mediated by the hydrazone linking spacer –NH–NCH–. Moreover, depending on the nature of their p-substituents, the arylethenyl pendent groups act either as electron-withdrawing (p-NO2, p-CN) or -releasing (p-Me) entities. We have shown that these respective electronic properties clearly influence the values of the redox potential in the cyclovoltammograms and the energies of the charge transfer bands in the electronic spectra. As a result, these compounds are proved to favour electronic delocalization along the conjugated chain. Finally, the polarized structure of this type of complexes, which endows them with solvatochromic properties, suggests that they are good candidates for non-linear optical studies. Further work will now be directed toward the determination of those properties for some selected dipolar organometallic hydrazone complexes reported here.
Experimental
General methods and materials
All operations were performed under inert atmosphere using standard vacuum/nitrogen line, Schlenk or syringe techniques with protection from light to avoid decomplexation of the CpFe+ moiety. The solvents were dried and distilled under nitrogen by standard methods prior to use. Microanalytical data were obtained by the Institut de Chimie de Rennes Microanalysis Service on a Thermo-Finigan Flash EA 1112 CHNS analyser. IR spectra were obtained with a Perkin Elmer Model 1600 FT-IR spectrophotometer. Electronic spectra were recorded in CH2Cl2 and DMSO solutions with a Spectronic Genesys 2 spectrophotometer. All the 1H NMR spectra were recorded in acetone-d6, unless otherwise stated, on multinuclear Bruker DPX 200 and Avance 400 Digital NMR Bruker spectrometers at 297 K; all chemical shifts are reported in parts per million (ppm) relative to internal tetramethylsilane (Me4Si), with the residual solvent proton resonance as internal standards. Coupling constants are given in Hertz (Hz).Electrochemical measurements were performed using a Radiometer Analytical model PGZ 100 All-in-One potentiostat, using a standard three-electrode setup with a vitreous carbon (in DMF) or Pt (in CH2Cl2) working electrode, platinum wire auxiliary electrode and Ag/AgCl as the reference electrode. DMF and CH2Cl2 solutions were 1.0 mM of the compound under study and 0.1 M of the supporting electrolyte n-Bu4N+PF6−. The Cp2Fe0/+ couple, located at 0.560 and 0.445 V under those conditions in DMF and CH2Cl2, respectively, was used as an internal reference for the potential measurements. E½ is defined as equal to (Epa+Epc)/2, where Epa and Epc are the anodic and cathodic peak potentials, respectively.
Chromatography columns (typically ca. 20 cm in length and ca. 2 cm in diameter) were packed with silica gel (Aldrich, 60 Å). Melting points were determined in evacuated capillaries and were not corrected.
The 4-(p-R-benzyl)triphenylphosphonium bromides (R=NO2, CN, Me) were prepared by a procedure similar to that described in ref. 50. 2-(1′-Formylferrocenyl)-1,3-dioxolane (2),21 1-formyl-1′-(E)-(p-nitrostyryl)ferrocene (5)17 and the hydrazine complexes [CpFe(η6-p-RC6H4NHNH2)]+PF6− (R=H, 7; Me, 8 and MeO, 9;26a R=Cl, 1013b) were prepared according to published procedures. 1-Formyl-1′-(E)-(p-cyanostyryl)ferrocene (6)17 was directly obtained as the desired pure E isomer using the protected/deprotected 1,3-dioxolane route described in ref. 21. Other chemicals were purchased from commercial sources and used as received.
Syntheses
°C. Anal. calcd for C22H22FeO2
(%): C, 70.60; H, 5.93; found: C, 70.75; H, 5.73. 1H NMR (400 MHz, CD3COCD3)
δ: 2.32 (s, 3H, CH3), 3.86–4.01 (m, 4H, CH2–CH2), 4.41 (t, 2H, C5H4, JH–H=1.8 Hz), 4.66 (t, 2H, C5H4, JH–H=1.8 Hz), 4.67 (t, 2H, C5H4, JH–H=1.8 Hz), 4.79 (t, 2H, C5H4, JH–H=1.8 Hz), 5.68 (s, 1H, O–CH–O), 6.78 (d, 1H, CHCH, JH–H=17 Hz), 6.93 (d, 1H, CHCH, JH–H=16 Hz), 7.16 (d, 2H, Ph, JH–H=7.3 Hz), 7.39 (d, 2H, Ph, JH–H=7.7 Hz). IR (KBr pellets)
νmax/cm−1: 3087w, 3014vw (CH), 2957w, 2917vw, 2841w, 2753vw (CH), 1664m (CC), 1091s, 1029s (C–O).
°C. Anal. calcd for C20H18FeO (%): C, 72.75; H, 5.49; found: C, 72.97; H, 5.56. 1H NMR (400 MHz, CD3COCD3)
δ: 2.32 (s, 3H, CH3), 4.41 (t, 2H, C5H4, JH–H=1.8 Hz), 4.62 (t, 2H, C5H4, JH–H=1.8 Hz), 4.67 (t, 2H, C5H4, JH–H=1.8 Hz), 4.78 (t, 2H, C5H4, JH–H=1.8 Hz), 6.82 (d, 1H, CHCH, JH–H=17 Hz), 6.87 (d, 1H, CHCH, JH–H=17 Hz), 7.17 (d, 2H, Ph, JH–H=8.1 Hz), 7.40 (d, 2H, Ph, JH–H=8.1 Hz), 9.91 (s, 1H, CHO). 13C{1H} NMR (100 MHz, CDCl3)
δ: 20.3 (CH3), 67.9 (C5H4), 70.2 (C5H4), 70.3 (C5H4), 73.9 (C5H4), 80.5 (q-C5H4), 85.7 (q-C5H4), 124.2 (CHCH), 126.0 (Ph), 127.7 (CHCH), 129.2 (Ph), 135.0 (q-Ph), 136.8 (q-Ph), 192.7 (CHO). IR (KBr pellets)
νmax/cm−1: 3090w, 3078w, 3045vw, 3022vw (CH), 2924vw, 2889w, 2856vw (CH), 1679s (HCO), 1663m (CC). UV-vis (CH2Cl2)
λmax/nm (Log ε/dm3 mol−1 cm−1): 263 (4.25), 318 (4.33), 366 (3.38), 464 (3.13); (DMSO): 264 (4.28), 320 (4.41), 374 (3.19), 474 (3.08). CV E1/2/V vs. Ag/AgCl (ΔEp/mV), v=0.1 V s−1: DMF (working electrode: vitreous carbon) 0.813 (119); CH2Cl2
(working electrode: Pt) 0.737 (196).
=H, 7; Me, 8; MeO, 9; Cl, 10) and 1-formyl-1′-(E)-(p-R′-styryl)ferrocene (R′=Me, 4, NO2, 5; CN, 6) was dissolved in 5 cm3 ethanol containing 5 drops of concentrated acetic acid. The solution was refluxed for 5 h, allowed to stand at room temperature and then at −30°C overnight (12 h). The precipitate was filtered off, washed first with cold EtOH and then with 5 cm3 of diethyl ether, then dried under vacuum. Crystallization from saturated dichloromethane solutions by slow diffusion of diethyl ether at room temperature provided the products as crystalline solids. Color, melting point and elemental analyses of the new hydrazone compounds 11–16 so prepared are given in Table 3. IR, UV-vis and 1H NMR data are gathered in Table 4.
°C overnight (12 h). The precipitate was filtered off and dissolved in 2 cm3 of dichloromethane. The solution was filtered through Celite and the filtrate was concentrated to half of its volume, then layered with an equivalent amount of diethyl ether to yield the products as crystalline solids. Color, melting point and elemental analyses of the new hydrazone compounds 17–20 so prepared are given in Table 3. IR, UV-vis and 1H NMR data are gathered in Table 4.
X-Ray Crystallography
Single crystals of (E)-[CpFe(η6-p-MeC6H4)–NHNCH–(η5-C5H4)Fe(η5-C5H4)–CHCH–C6H4-p-Me]+PF6−
(16) and of [{CpFe(η6-p-MeOC6H4)–NHNCH–(η5-C5H4)}2Fe]2+(PF6−)2
(19) were obtained by slow diffusion of diethyl ether into the corresponding concentrated CH2Cl2 solutions of complexes 16 and 19 at room temperature.
A single crystal of complex 16 was mounted on a glass fiber in a random orientation. Data collection was performed at room temperature on a Siemens Smart CCD diffractometer using graphite-monochromated Mo-Kα radiation (λ=0.71073 Å), using 0.3° of separation between frames and 30 s per frame. Space group assignments are based on systematic absences, E statistics and successful refinement of the structure. The structure was solved by direct methods with the aid of successful difference Fourier maps and was refined using the SHELXTL 5.1 software package.51 All non-hydrogen were refined anisotropically. Hydrogen atoms were assigned to ideal positions and refined using a riding model. The diffraction frames were integrated using the SAINT52 package and corrected for absorption with SADABS.53
In the case of complex 19, the data collection was made on a Bruker SMART APEX diffractometer, using 0.3° of separation between frames and 10 s per frame. The structure was solved and refined using SHELXS9754 and SHELXL97,54 respectively, both included in the WinGX55 software package.
The final R indices as well as further details concerning the resolution and refinement of the crystal structures of 16 and 19 are presented in Table 7.†
| 16 | 19 | |
|---|---|---|
| Empirical formula | C32H31F6Fe2N2P | C18H18F6Fe1.5N2OP |
| FW/g mol−1 | 700.26 | 507.09 |
| T/K | 293(2) | 293(2) |
| Crystal system | Triclinic | Orthorhombic |
| Space group | P-1 | Pbca |
| a/Å | 10.612(3) | 12.5730(11) |
| b/Å | 11.147(3) | 16.5448(14) |
| c/Å | 13.521(4) | 19.0521(16) |
| α/° | 85.155(7) | 90 |
| β/° | 74.476(7) | 90 |
| γ/° | 77.999(7) | 90 |
| U/Å3 | 1506.8(8) | 3963.2(6) |
| Z | 2 | 8 |
| μ/mm−1 | 1.079 | 1.260 |
| λ(Mo Kα)/Å | 0.71073 | 0.71073 |
| No. total reflect. | 8369 | 27421 |
| No. unique reflect. | 5129 | 4559 |
| R int | 0.0332 | 0.0944 |
|
R
1
[I>2σ(I)] |
0.0651 | 0.0442 |
|
wR
2
[I>2σ(I)] |
0.1192 | 0.0951 |
| R 1 (all data) | 0.0949 | 0.1291 |
| wR 2 (all data) | 0.1271 | 0.0991 |
Acknowledgements
We thank Dr. Andrés Vega (Santiago de Chile) for helpful assistance in the structure determination of 19. We greatly appreciate financial support for this work from the Fondo Nacional de Desarrollo Científico y Tecnológico, FONDECYT (Chile), Grant N° 1010318 (D. C., C. M.), to the Programme International de Coopération Scientifique N° 922 CNRS (France)–CONICYT (Chile and the CNRS-CONICYT Project N° 14531) (C. M., D. C., J.-R. H.), and to the Vicerrectoría de Investigación y Estudios Avanzados, Pontificia Universidad Católica de Valparaíso, Valparaíso, Chile.References
- Ferrocenes: Homogenous Catalysis, Organic Synthesis, Materials Science, eds. A. Togni and T. Hayashi, Wiley-VCH, Weinheim, 1995 Search PubMed.
- (a) D. Astruc, Pure Appl. Chem., 2003, 75, 461 Search PubMed; (b) D. Astruc, J.-C. Blais, M.-C. Daniel, V. Martinez, S. Nlate and J. Ruiz, Macromol. Symp., 2003, 196, 1 CrossRef CAS; (c) B. Alonso, E. Alonso, D. Astruc, J.-C. Blais, L. Djakovitch, J.-L. Fillaut, S. Nlate, F. Moulines, S. Rigaut, J. Ruiz and C. Valerio, Adv. Dendritic Macromol., 2002, 5, 89 Search PubMed.
- (a) R. D. A. Hudson, J. Organomet. Chem., 2001, 637–639, 47 CrossRef CAS; (b) P. Nguyen, P. Gómez-Elipe and I. Manners, Chem. Rev., 1999, 99, 1515 CrossRef CAS; (c) A. S. Abd-El-Aziz, Macromol. Rapid Commun., 2002, 23, 995 CrossRef CAS.
- J. S. Miller and A. J. Epstein, in Research Frontiers in Magnetochemistry, ed. C. J. O'Connor, World Scientific, Singapore, 1993, pp. 283–302 and references therein Search PubMed.
- (a) S. Barlow and S. R. Marder, Chem. Commun., 2000, 1555 RSC; (b) H. S. Nalwa, Appl. Organomet. Chem., 1991, 5, 349 CrossRef CAS.
- For an overview, see: (a) S. Di Bella, Chem. Soc. Rev., 2001, 30, 355 RSC; (b) J. Heck, S. Dabek, T. Meyer-Friedrichsen and H. Wong, Coord. Chem. Rev., 1999, 190–192, 1217 CrossRef CAS; (c) I. R. Whittall, A. M. McDonagh, M. G. Humphrey and M. Samoc, Adv. Organomet. Chem., 1998, 42, 291; (d) N. J. Long, Angew. Chem., Int. Ed. Engl., 1995, 34, 21 CrossRef CAS; (e) D. R. Kanis, M. A. Ratner and T. J. Marks, Chem. Rev., 1994, 94, 195 CrossRef CAS.
- A. N. Nesmeyanov, E. G. Perevalova, S. P. Gubin, K. I. Granberg and A. G. Kozlovsky, Tetrahedron Lett., 1966, 7, 2381 CrossRef.
- (a) D. Astruc, S. Nlate and J. Ruiz, in Modern Arene Chemistry, ed. D. Astruc, Wiley-VCH, Weinheim, 2002, ch. 12, pp. 400–434 Search PubMed; (b) A. S. Abd-El-Aziz and S. Bernardin, Coord. Chem. Rev., 2000, 203, 219 CrossRef CAS; (c) D. Astruc, Top. Curr. Chem., 1991, 160, 47; (d) D. Astruc, Tetrahedron, 1983, 39, 4027 CrossRef CAS.
- D. Astruc, Acc. Chem. Res., 1997, 30, 383 CrossRef CAS.
- D. Astruc, Chem. Rev., 1988, 88, 1189 CrossRef CAS.
- (a) W. A. Hendrickson and M. C. Palazzotto, Photosensitive Metal-Organic Systems, eds. C. Kutal and N. Serpone, Advanced Chemistry Series 238, American Chemical Society, Washington, DC, 1993, pp. 411–432 Search PubMed; (b) D. R. Chrisope, K. M. Park and G. B. Schuster, J. Am. Chem. Soc., 1989, 111, 6195 CrossRef CAS; (c) D. R. Chrisope and G. B. Schuster, Organometallics, 1989, 8, 2737 CrossRef CAS.
- C. Lambert, W. Gaschler, M. Zabel, R. Matschiner and R. Wortmann, J. Organomet. Chem., 1999, 592, 109 CrossRef CAS.
- (a) C. Manzur, L. Millán, M. Fuentealba, J.-R. Hamon and D. Carrillo, Tetrahedron Lett., 2000, 41, 3615 CrossRef CAS; (b) C. Manzur, M. Fuentealba, L. Millán, F. Gajardo, D. Carrillo, J. A. Mata, S. Sinbandhit, P. Hamon, J.-R. Hamon, S. Kahlal and J.-Y. Saillard, New J. Chem., 2002, 26, 213 RSC; (c) C. Manzur, M. Fuentealba, D. Carrillo, D. Boys and J.-R. Hamon, Bol. Soc. Chil. Quim., 2001, 46, 409 Search PubMed; (d) C. Manzur, M. Fuentealba, L. Millán, F. Gajardo, M. T. Garland, R. Baggio, J. A. Mata, J.-R. Hamon and D. Carrillo, J. Organomet. Chem., 2002, 660, 71 CrossRef CAS; (e) A. Trujillo, M. Fuentealba, C. Manzur, D. Carrillo and J.-R. Hamon, J. Organomet. Chem., 2003, 681, 150 CrossRef CAS.
- (a) C. Serbutoviez, C. Bosshard, G. Knöpfle, P. Weiss, P. Prêtre, P. Günter, K. Schenk, E. Solari and G. Chapuis, Chem. Mater., 1995, 7, 1198 CrossRef CAS; (b) M. S. Wong, U. Meier, F. Pan, V. Gramlich, C. Bosshard and P. Günter, Adv. Mater., 1996, 8, 416 CAS.
- (a) A. S. Abd-El-Aziz, E. K. Todd, R. M. Okasha and T. E. Wood, Macromol. Rapid Commun., 2002, 23, 743 CrossRef CAS; (b) A. S. Abd-El-Aziz, E. K. Todd and R. M. Okasha, Macromol. Symp., 2003, 196, 77 CrossRef CAS.
- O. Briel, K. Sünkel, I. Krossing, H. Noth, E. Schmalzlin, K. Meerholz, C. Brauchle and W. Beck, Eur. J. Inorg. Chem., 1999, 483 CrossRef CAS.
- J. A. Mata, E. Peris, R. Llusar, S. Uriel, M. P. Cifuentes, M. G. Humphrey, M. Samoc and B. Luther-Davies, Eur. J. Inorg. Chem., 2001, 2113 CrossRef CAS.
- J. A. Mata and E. Peris, Inorg. Chim. Acta, 2003, 343, 175 CrossRef CAS.
- S. K. Hurst, M. G. Humphrey, J. P. Morrall, M. P. Cifuentes, M. Samoc, B. Luther-Davies, G. A. Heath and A. C. Willis, J. Organomet. Chem., 2003, 670, 56 CrossRef CAS.
- (a) J. C. Calabrese, L.-T. Cheng, J. C. Green, S. R. Marder and W. Tam, J. Am. Chem. Soc., 1991, 113, 7227 CrossRef CAS; (b) H. E. Bunting, M. L. H. Green, S. R. Marder, M. E. Thompson, D. Bloor, P. V. Kolinsky and R. J. Jones, Polyhedron, 1992, 11, 1489 CrossRef CAS.
- J. Chiffre, F. Averseng, G. G. A. Balavoine, J.-C. Daran, G. Iftime, P. G. Lacroix, E. Manoury and K. Nakatani, Eur. J. Inorg. Chem., 2001, 2221 CrossRef CAS.
- (a) Modifications in the work -up of 1,1′-ferrocenedicarboxaldehyde22b have been carried out; see Experimental section for details; (b) G. G. A. Balavoine, G. Doisneau and T. Fillebeen-Khan, J. Organomet. Chem., 1991, 412, 381 CrossRef CAS.
- D. Astruc, Electron Transfer and Radical Reactions in Transition-Metal Chemistry, VCH, New York, 1995, ch. 2, pp. 89–195 Search PubMed.
- C. Manzur, L. Millán, W. Figueroa, D. Boys, J.-R. Hamon and D. Carrillo, Organometallics, 2003, 22, 153 CrossRef CAS.
- H. A. Trujillo, C. M. Casado, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 1999, 121, 5674 CrossRef CAS.
- (a) C. Manzur, E. Baeza, L. Millán, M. Fuentealba, P. Hamon, J.-R. Hamon, D. Boys and D. Carrillo, J. Organomet. Chem., 2000, 608, 126 CrossRef CAS; (b) W. Figueroa, M. Fuentealba, C. Manzur, D. Carrillo, J. A. Mata and J.-R. Hamon, J. Chil. Chim. Soc., 2003, 48, 75 Search PubMed.
- (a) S. Marcen, M. V. Jiménez, I. T. Dobrinovich, F. J. Lahoz, L. A. Oro, J. Ruiz and D. Astruc, Organometallics, 2002, 21, 326 CrossRef CAS; (b) Y. Ishii, M. Kawaguchi, Y. Ishino, T. Aoki and M. Hidai, Organometallics, 1994, 13, 5062 CrossRef CAS; (c) S. Subramanian, L. Wang and M. J. Zaworotko, Organometallics, 1993, 12, 310 CrossRef CAS; (d) J.-L. Fillaut, R. Boese and D. Astruc, Synlett, 1992, 55 CrossRef CAS; (e) K. A. Abboud, S. H. Simonsen, A. Piorko and R. G. Sutherland, Acta Crystallogr., Sect. C, 1991, 47, 860 CrossRef.
- For exemples of X-ray crystal structures of [CpFe(η6-arene)]+ complexes, see: (a) J.-R. Hamon, J.-Y. Saillard, A. Le Beuze, M. J. McGlinchey and D. Astruc, J. Am. Chem. Soc., 1982, 104, 7549 CrossRef CAS; (b) See also refs 11, 13b–d, 24, 26, 27 and 29.
- P. J. Dyson, M. C. Grossel, N. Shrinivasan, T. Vine, T. Welton, D. J. Williams, A. J. P. White and T. Zigras, J. Chem. Soc., Dalton Trans., 1997, 3465 RSC.
- For structurally characterized bis-substituted ferrocene-based π-bridged systems, see, for example, refs 16–21.
- For a reference gathering a large number of interatomic and metal-ligand distances obtained from the Cambridge Crystallographic Data Base Centre, see: A. G. Orpen, L. Brammer, F. H. Allen, D. Kennard, D. G. Watson and R. Taylor, J. Chem. Soc., Dalton Trans., 1989, S1 Search PubMed.
- The deconvolution of the lower energy absorption band gave rise to a splitting into two independent bands whose λmax are observed in the 479–483 nm range. We also expect d-d transitions, due to both the cationic mixed sandwich (ref. 33) and the ferrocenyl unit (ref. 34), to be present in the visible part of the spectrum. However, we expect these to be very weak compared to the two charge transfer transitions.
- (a) W. H. Morrison, E. Y. Ho and D. N. Hendrickson, Inorg. Chem., 1975, 14, 500 CrossRef CAS; (b) J.-R. Hamon, D. Astruc and P. Michaud, J. Am. Chem. Soc., 1981, 103, 758 CrossRef CAS.
- (a) Y. S. Sohn, D. N. Hendrickson and H. B. Gray, J. Am. Chem. Soc., 1971, 93, 3603 CrossRef; (b) Y. Yamaguchi and C. Kutal, Inorg. Chem., 1999, 38, 4861 CrossRef CAS.
- S. Barlow, H. E. Bunting, C. Ringham, J. C. Green, G. U. Bublitz, S. G. J. Boxer, J. W. Perry and S. R. Marder, J. Am. Chem. Soc., 1999, 121, 3715 CrossRef CAS.
- For neutral chromophores see, for example: (a) J. Mata, S. Uriel, E. Peris, R. Llusar, S. Houbrechts and A. Persoons, J. Organomet. Chem., 1998, 562, 197 CrossRef CAS; (b) G. G. A. Balavoine, J.-C. Daran, G. Iftime, P. G. Lacroix, E. Manoury, J. A. Delaire, I. Maltey-Fanton, K. Nakatani and S. Di Bella, Organometallics, 1999, 18, 21 CrossRef CAS; (c) J. A. Mata, E. Falomir, R. Llusar and E. Peris, J. Organomet. Chem., 2000, 616, 80 CrossRef CAS; (d) J. A. Mata, S. Uriel, R. Llusar and E. Peris, Organometallics, 2000, 19, 3797 CrossRef CAS; (e) J. A. Mata, E. Peris, I. Asselberghs, R. Van-Boxel and A. Persoons, New J. Chem., 2001, 25, 299 RSC; (f) I. Janowska, J. Zakrzewski, K. Nakatani, J. A. Delaire, M. Palusiak, M. Walak and H. Scholl, J. Organomet. Chem., 2003, 675, 35 CAS.
- For cationic chromophores see, for example: (a) V. Alain, A. Fort, M. Barzoukas, C.-T. Chen, M. Blanchard-Desce, S. R. Marder and J. W. Perry, Inorg. Chim. Acta, 1996, 242, 43 CrossRef CAS; (b) T. Farrell, T. Meyer-Friedrichsen, M. Malessa, D. Haase, W. Saak, I. Asselberghs, K. Wostyn, K. Clays, A. Persoons, J. Heck and A. R. Manning, J. Chem. Soc., Dalton Trans., 2001, 29 RSC; (c) H. Wong, T. Meyer-Friedrichsen, T. Farrell, C. Mecker and J. Heck, Eur. J. Inorg. Chem., 2000, 631 CrossRef; (d) B. J. Coe, L. A. Jones, J. A. Harris, B. S. Brunschwig, I. Asselberghs, K. Clays and A. Persoons, J. Am. Chem. Soc., 2003, 125, 862 CrossRef CAS.
- R. P. Hsung, C. E. D. Chidsey and L. R. Sita, Organometallics, 1995, 14, 4808 CrossRef CAS.
- (a) A. S. Abd-El-Aziz, T. H. Afifi, W. R. Budakowski, K. J. Friesen and E. K. Todd, Macromolecules, 2002, 35, 8929 CrossRef CAS; (b) A. S. Abd-El-Aziz, E. K. Todd, R. M. Okasha and T. H. Afifi, Macromol. Symp., 2003, 196, 89 CrossRef CAS.
- (a) C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, 2nd edn., VCH, Wenheim, 1988 Search PubMed; (b) C. Reichardt, Chem. Rev., 1994, 94, 2319 CrossRef CAS and references therein.
- A hypsochromic shift has also been reported for unusual neutral chromophores: P. D. Beer and H. Sikanyika, Polyhedron, 1990, 9, 1091 Search PubMed.
- P. Zanello, in Ferrocenes: Homogenous Catalysis, Organic Synthesis, Materials Science, eds. A. Togni and T. Hayashi, Wiley-VCH, Weinheim, 1995, ch. 7, pp. 317–430 Search PubMed.
- The redox potentials of compounds 11–20 and those of their counterparts prepared in ref. 13b are compared relative to the ferrocene/ferricinium couple used in both cases as internal reference.
- The benzylic N–H group is indeed strongly activated by both the electron-withdrawing organometallic moiety CpFe+ and the imine functionality. Therefore, the thermally stable zwitterion [CpFe+(p-RC6H4)N−–NCH(C5H4)Fe(C5H4R′)] can be easily formed by deprotonation in the electrochemical medium. No attempts were made to characterize their corresponding non-deprotonated precursors 11–20 by CV.
- This fully irreversible wave could indeed be attributed to the 2-electron reduction of the C–Cl functionality, following the ECE mechanism for the reduction of Ar–X into Ar˙ and X− (ref. 46). Its integrated area is twice that of the wave at −2.44 V.
- D. G. Peters, in Organic Electrochemistry, eds. H. Lund and O. Hammerich, 4th edn., Marcel Dekker, New York, 2001, ch. 8, pp. 341–377 Search PubMed.
- B. S. Jensen and V. D. Parker, J. Chem. Soc., Chem. Commun., 1974, 367 RSC.
- H. Lund, in Organic Electrochemistry, eds. H. Lund and O. Hammerich, 4th edn., Marcel Dekker, New York, 2001, ch. 9, pp. 379–409 Search PubMed.
- For detailed mechanistic investigations on the reduction of nitroaromatic compounds see: (a) A. Darchen and C. Moinet, J. Electroanal. Chem., 1976, 68, 173 CrossRef CAS; (b) A. Darchen and C. Moinet, J. Chem. Soc., Chem. Commun., 1976, 820 Search PubMed; (c) A. Darchen and C. Moinet, J. Electroanal. Chem., 1977, 81, 78.
- R. Ketcham, D. Jambotkar and L. Martinelli, J. Org. Chem., 1962, 27, 4666 CAS.
- G. M. Sheldrick, SHELXTL, version 5.1, Bruker AXS, Inc., Madison, WI, USA, 1997 Search PubMed.
- SAINT, version 5.0, Bruker Analytical X-Ray Systems, Madison, WI, USA, 1998 Search PubMed.
- G. M. Sheldrick, SADABS Empirical Absorption Program, University of Göttingen, Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELXS-97, Program for solution of crystal structures (release 97-2), University of Göttingen, Germany, 1997 Search PubMed; G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures (release 97-2), University of Göttingen, Germany, 1997 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef.
Footnote |
| † CCDC reference numbers 188753 for 16 and 209331 for 19. See http://www.rsc.org/suppdata/nj/b3/b308626g/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2004 |