Racemic vanadium(V) oxo monoperoxo complexes with two achiral bidentate heteroligands. Synthesis, characterization, crystal structure and stereochemistry of K[VO(O2)(ox)(bpy)]·3H2O and Pr4N[VO(O2)(ox)(phen)]
Jozef
Tatiersky
*a,
Peter
Schwendt
a,
Jaromír
Marek
b and
Michal
Sivák
a
aDepartment of Inorganic Chemistry, Faculty of Natural Sciences, Comenius University, Mlynská dolina, 842 15, Bratislava 4, Slovak Republic. E-mail: tatiersky@fns.uniba.sk; Fax: +421 2 6029 6273; Tel: +421 2 6029 6328
bLaboratory of Functional Genomics and Proteomics, Faculty of Natural Sciences, Masaryk University, Kotlářská 2, 611 37, Brno, Czech Republic
First published on 14th November 2003
Abstract
Two new racemic monoperoxo complexes of vanadium(V), K[VO(O2)(ox)(bpy)]·3H2O (1) and Pr4N[VO(O2)(ox)(phen)]
(2)
[bpy![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =2,2′-bipyridine, phen=1,10-phenanthroline, ox=oxalate(2−) and Pr4N=tetra(n-propyl)ammonium(1+)], were synthesized. The solid complexes were characterized by X-ray structure analysis and IR and Raman spectroscopies. The coordination polyhedron of vanadium is a distorted pentagonal bipyramid. The five equatorial positions are occupied by the η2-peroxo oxygen atoms, two oxygen atoms of the ox ligand and one nitrogen atom of bpy (resp. phen). The oxo ligand and the second nitrogen atom of bpy (resp. phen) are in axial positions. In each structure, the intermolecular distance, 3.5 Å, between the pairs of parallel bpy or phen ligands indicates a π-π interaction between the aromatic rings. The UV-vis spectra of aqueous solutions exhibit characteristic peroxo-to-vanadium CT bands at 422 nm (ε=32 m2 mol−1) for 1 and 428 nm (ε=29.4 m2 mol−1) for 2. From the 51V NMR measurements, the isomerization based on a mutual ox↔bpy or ox↔phen position change does not occur in aqueous solutions. The structure of both complex anions is maintained at 295 K for about 6 h after dissolution in water.
=2,2′-bipyridine, phen=1,10-phenanthroline, ox=oxalate(2−) and Pr4N=tetra(n-propyl)ammonium(1+)], were synthesized. The solid complexes were characterized by X-ray structure analysis and IR and Raman spectroscopies. The coordination polyhedron of vanadium is a distorted pentagonal bipyramid. The five equatorial positions are occupied by the η2-peroxo oxygen atoms, two oxygen atoms of the ox ligand and one nitrogen atom of bpy (resp. phen). The oxo ligand and the second nitrogen atom of bpy (resp. phen) are in axial positions. In each structure, the intermolecular distance, 3.5 Å, between the pairs of parallel bpy or phen ligands indicates a π-π interaction between the aromatic rings. The UV-vis spectra of aqueous solutions exhibit characteristic peroxo-to-vanadium CT bands at 422 nm (ε=32 m2 mol−1) for 1 and 428 nm (ε=29.4 m2 mol−1) for 2. From the 51V NMR measurements, the isomerization based on a mutual ox↔bpy or ox↔phen position change does not occur in aqueous solutions. The structure of both complex anions is maintained at 295 K for about 6 h after dissolution in water.
Introduction
In the last decades, the discovery of the biological activity of vanadium compounds has initiated an intensive development in the bioinorganic chemistry of vanadium. The compounds of interest include vanadium(V) complexes with the peroxovanadium(V) moiety, VO(O2)+, which was found to be formed, in the presence of hydrogen peroxide, in the active site of vanadium haloperoxidases: enzymes isolated from marine algae (Ascophyllum nodosum) or microscopic funghi (Curvularia inaequalis).1,2 The insulin mimetic behaviour of vanadium(V) peroxo complexes, their antitumour activity3–5 and the ability to inhibit some enzymes, such as alkaline phosphatase in green scrabs (Scylla serrata),6 were also investigated. Peroxo complexes also play an important role as oxidants of organic substrates, as in the epoxidation and hydroxylation of hydrocarbons7 or in the enantioselective oxidation of sulfides.8–10 In the latter reaction, the monoperoxo but not the diperoxo complexes are assumed to be the active species.8The completion of the coordination sphere of the vanadium in the VO(O2)+ monoperoxovanadium moiety by (a) heteroligand(s) with different denticity and providing different donor sets of oxygen and nitrogen atoms can result in the formation of a great variety of structural isomers. The study of such complexes is of great interest not only from the stereochemical aspect, but it can also lead to better understanding of their reactivity. The vanadium atoms in oxo peroxo complexes are either six-coordinated, with a distorted pentagonal pyramidal geometry, or seven-coordinated, with a distorted pentagonal bipyramidal arrangement of the donor atoms. The coordination number six was observed in structures of several dinuclear complexes with α-hydroxycarboxylates11 and in the mononuclear glycylglycine complex, Et4N[VO(O2)(glygly)]·1.58H2O.12 Nevertheless, in the latter complex, a weak interaction between the vanadium atom in one [VO(O2)(glygly)]− anion and the peroxo oxygen atom of the neighbouring anion results in the formation of pairs of complex anions. The coordination numbers of two vanadium atoms in a pair, with “neighbouring” peroxo oxygen in the second apical position, can thus also be considered to be seven. A similar completion of the coordination sphere was also observed in structures of the phosphato peroxo complexes of vanadium(V).13
The vanadium atoms in the monoperoxo complexes are chirotopic. Generally, these complexes with achiral or racemic heteroligands crystallize as: (i) racemic compounds such as [VO(O2)(phen)(H2O)2]Cl·0.38H2O14 or K[VO(O2)(DL-cmhist)]·H2O,15 (ii) racemic conglomerates though the peroxo iminodiacetato complex, NH4[VO(O2)(ida)], is the only one known so far16 or (iii) dimeric meso compounds, mainly with α-hydroxycarboxylates, as in (NBu4)2[V2O2(O2)2(C2H2O3)2]·H2O,17 (NBu4)2[V2O2(O2)2(D-lact)(L-lact)]·2H2O11,18 or Et4N[VO(O2)(glygly)]·1.58H2O.12
The syntheses of complexes with chiral non-racemic heteroligands result in the formation of chiral non-racemic compounds with a structure in a chiral space group (Sohncke groups) as, for example, the complexes (NBu4)2[V2O2(O2)2(L-lact)2]·2H2O11 and K2[{VO(O2)(L-tartH2)}2(μ-H2O)]·5H2O.19 The native and peroxo forms of the vanadium haloperoxidase also crystallize as chiral non-racemic compounds, because of the natural chiral environment in which they are formed.2,20,21
In all structures of vanadium(V) oxo peroxo complexes determined so far, the oxo and peroxo ligands are in a cis arrangement. A further structural feature is the usually observed trans influence, which is manisfested by elongation of the bond length between the vanadium atom and donor atom in the position trans to the oxo ligand, when compared with the bond lengths between the vanadium atom and donor atoms in cis positions. A similar trans influence can also be observed on the bond length between the vanadium atom and donor atom in the position trans to the peroxo ligand (Fig. 1).
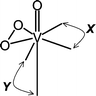 | ||
| Fig. 1 General structural formula for the vanadium(V) oxo peroxo complexes with two bidentate heteroligands X and Y. The equatorial plane is defined by the η2-peroxo oxygen atoms, two donor atoms of X and one of Y. | ||
In vanadium(V) oxo peroxo complexes with two bidentate heteroligands X and Y, [VO(O2)(X)(Y)], the coordination polyhedron of the vanadium atom is distorted pentagonal bipyramid. The donor atoms of a heteroligand X occupy two equatorial positions (EEP), while those of Y are in equatorial and apical positions (EAP).
The ligands X and Y are mostly σ-donors or both σ-donors and π-acceptors providing OO, NO or NN donor sets and forming on coordination two five-membered chelate rings. The preferred binding of one of the two competing bidentate heteroligands in two particular (EEP or EAP) sites of the four “free” coordination sites of the vanadium atom in [VO(O2)(X)(Y)] complexes seems to be their characteristic stereochemical feature. However, the factors determining this phenomenon still remain unclear.
We present here a study of vanadium(V) oxo monoperoxo complexes with two bidentate heteroligands that differ in donor sets and charges: ox-bpy and ox-phen. The molecular and crystal structures, IR and Raman spectra, as well as the 51V NMR spectra of aqueous solutions of the prepared complexes, and of the corresponding reaction solutions, are discussed.
Experimental
Materials
2,2′-Bipyridine (bpy), 1,10-phenanthroline monohydrate (phen·H2O), oxalic acid dihydrate (H2ox·2H2O), hydrogen peroxide, acetonitrile, dichloromethane and tetrapropylammonium hydroxide were used as supplied by Merck, Lachema, or Slavus. KVO3 was prepared according to ref. 22 while V2O5 was prepared by thermal decomposition of purified NH4VO3 at 773 K.Measurements
The IR spectra in nujol mulls were registered on a FT IR Nicolet Magna 750 spectrometer. The Raman spectra were registered on a FT RA model FRA 106/S connected to a FT IR spectrometer FS 55 Equinox (Bruker); a Nd:YAG laser (1.064 µm, 350–500 mW) was used.The 51V NMR spectra of aqueous solutions were registered at room temperature or at 278 K on a Varian Gemini 2000 spectrometer (B0=7.05 T; 78.9 MHz operating frequency). The chemical shifts are related to VOCl3 as external standard. The samples of reaction solutions for 51V NMR measurements were taken after preparation and before standing to crystallize.
The UV-vis spectra of 0.02 mol L−1 aqueous solutions of 1 and 2 were measured on a Hewlett Packard 84-52A spectrophotometer: 8 min after dissolution, at 295 K and in a 1 mm cell.
Carbon, hydrogen and nitrogen were determined on a Carlo Erba analyzer. Vanadium was determined gravimetrically as KVO3 in 1 or as V2O5 in 2, after annealing the sample in a Pt crucible at 773 K and comfirming their identity and purity by diffraction patterns registered on a Philips PW 1050 diffractometer using CuKα radiation.
X-Ray crystallographic studies
The diffraction data were collected with a KM4CCD diffractometer with a four-circle area detector (KUMA Diffraction, Poland) and equipped with an Oxford Cryostream Cooler (Oxford Cryosystems, UK). MoKα radiation was used in all measurements. The ω scan technique with different κ and φ offsets for covering a whole independent part of the reflections in the 2°–25° θ range was performed. The cell parameters were refined from all strong reflections (>1000 pulses). The data reductions were carried out using the CrysAlis RED (Oxford Diffraction, UK) program. The structures were determined by SHELXS-9723 and refined by SHELXL-97,24 the data for publication were prepared by SHELXL and PARST,25 and the figures by the ORTEP-III26a and STRUPLO26b programs.†=436.29, monoclinic, a=9.6777(5), b=13.8879(6), c=12.7353(6)
Å, β=97.606(5)°, V=1696.60(14)
Å3, T=150(2) K, space group P21/c
(no. 14), Z=4, μ=0.887 mm−1, 9593 reflections measured, 2968 unique (Rint=0.0248), which were used in all calculations. The final wR(F2) was 0.0615 (all data).
=553.52, monoclinic, a=29.5519(12), b=10.0503(5), c=19.3160(9)
Å, β=112.005(4)°, V=5319.0(4)
Å3, T=120(2) K, space group C2/c
(no. 15), Z=8, μ=0.422 mm−1, 14505 reflections measured, 4654 unique (Rint=0.0401), which were used in all calculations. The final wR(F2) was 0.1021 (all data).
Syntheses
=3.3), which was kept in a Petri dish in a refrigerator at 278 K. The red crystals of 1, which formed within 3 days, were filtered off and washed with a small amount of cooled water. The identical product was also obtained on crystallization from the filtrate. Anal. calcd. for C12H14KN2O10V: 33.04 C, 3.23 H, 6.42 N, 11.68 V; found: 33.40 C, 3.30 H, 6.71 N, 11.53 V. UV-vis: peroxo-to-vanadium CT band at 422 nm (ε=32 m2 mol−1).
=29.4 m2 mol−1).
Results and discussion
Crystal structures
In the structures of both complexes, the coordination polyhedron of the vanadium atom is a distorted pentagonal bipyramid as schematically shown in Fig. 1 for a monoperoxo complex with the general formula [VO(O2)(X)(Y)]. The donor atoms of the oxalato ligand in 1 and 2 occupy the EEP, while bpy (resp. phen) occupies the EAP (Figs. 2 and 3).![Molecular structure of [VO(O2)(ox)(bpy)]− with 50% probability ellipsoids. The hydrogen atoms are omitted. Selected bond lengths (Å) and angles (°): V(1)–O(1) 1.6030(13), V(1)–O(2) 1.8988(13), V(1)–O(3) 1.8648(13), V(1)–O(4) 2.0663(13), V(1)–O(5) 2.0272(13), V(1)–N(1) 2.1291(15), V(1)–N(2) 2.2583(15), O(2)–O(3) 1.4366(18), O(1)–V(1)–N(1) 93.93(6), O(1)–V(1)–N(2) 166.61(6), O(1)–V(1)–O(2) 99.74(7), O(1)–V(1)–O(3) 103.95(6), O(1)–V(1)–O(4) 94.49(6), O(1)–V(1)–O(5) 98.62(6), O(2)–O(3)–V(1) 68.82(7), O(3)–O(2)–V(1) 66.31(7).](/image/article/2004/NJ/b306090j/b306090j-f2.gif) | ||
| Fig. 2 Molecular structure of [VO(O2)(ox)(bpy)]− with 50% probability ellipsoids. The hydrogen atoms are omitted. Selected bond lengths (Å) and angles (°): V(1)–O(1) 1.6030(13), V(1)–O(2) 1.8988(13), V(1)–O(3) 1.8648(13), V(1)–O(4) 2.0663(13), V(1)–O(5) 2.0272(13), V(1)–N(1) 2.1291(15), V(1)–N(2) 2.2583(15), O(2)–O(3) 1.4366(18), O(1)–V(1)–N(1) 93.93(6), O(1)–V(1)–N(2) 166.61(6), O(1)–V(1)–O(2) 99.74(7), O(1)–V(1)–O(3) 103.95(6), O(1)–V(1)–O(4) 94.49(6), O(1)–V(1)–O(5) 98.62(6), O(2)–O(3)–V(1) 68.82(7), O(3)–O(2)–V(1) 66.31(7). | ||
![Molecular structure of [VO(O2)(ox)(phen)]− with 50% probability ellipsoids. The hydrogen atoms are omitted. Selected bond lengths (Å) and angles (°): V(1)–O(1) 1.6065(17), V(1)–O(2) 1.8569(18), V(1)–O(3) 1.8841(18), V(1)–O(4) 2.0084(18), V(1)–O(5) 2.0458(17), V(1)–N(1) 2.151(2), V(1)–N(2) 2.321(2), O(2)–O(3) 1.415(2), O(1)–V(1)–N(1) 93.92(8), O(1)–V(1)–N(2) 166.00(8), O(1)–V(1)–O(2) 104.68(9), O(1)–V(1)–O(3) 101.34(8), O(1)–V(1)–O(4) 102.22(8), O(1)–V(1)–O(5) 92.31(8), O(2)–O(3)–V(1) 66.75(10), O(3)–O(2)–V(1) 68.79(10).](/image/article/2004/NJ/b306090j/b306090j-f3.gif) | ||
| Fig. 3 Molecular structure of [VO(O2)(ox)(phen)]− with 50% probability ellipsoids. The hydrogen atoms are omitted. Selected bond lengths (Å) and angles (°): V(1)–O(1) 1.6065(17), V(1)–O(2) 1.8569(18), V(1)–O(3) 1.8841(18), V(1)–O(4) 2.0084(18), V(1)–O(5) 2.0458(17), V(1)–N(1) 2.151(2), V(1)–N(2) 2.321(2), O(2)–O(3) 1.415(2), O(1)–V(1)–N(1) 93.92(8), O(1)–V(1)–N(2) 166.00(8), O(1)–V(1)–O(2) 104.68(9), O(1)–V(1)–O(3) 101.34(8), O(1)–V(1)–O(4) 102.22(8), O(1)–V(1)–O(5) 92.31(8), O(2)–O(3)–V(1) 66.75(10), O(3)–O(2)–V(1) 68.79(10). | ||
The V(1)–O(1) distances, 1.6030 and 1.6065 Å, are within the range observed in structures of the majority of vanadium(V) monoperoxo complexes and are typical for doubly bonded oxygen atoms. The peroxo ligand is slightly asymmetrically coordinated to the vanadium atom. In the structures of 1 and 2, the above described trans influences can be observed: d[V(1)–N(1)]<d[V(1)–N(2)]
(for 1 and 2), d[V(1)–O(5)]<d[V(1)–O(4)]
(for 1) and d[V(1)–O(4)]<d[V(1)–O(5)]
(for 2). All O(1)–V(1)–Q bond angles (Q being the donor atoms in the pentagonal plane) are greater than 90° as a consequence of the deviation of the vanadium atom from the pentagonal plane towards the oxo oxygen atom by 0.2712(7)
Å for 1 and 0.2941(9)
Å for 2.
As follows from the centrosymmetric space groups, the complexes crystallize as racemic compounds. The pairs of vanadium polyhedra representing two enantiomers are mutually centrosymmetrically oriented as shown for 1 in Fig. 4.
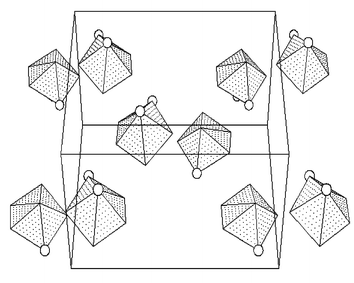 | ||
| Fig. 4 Packing of VN2O5 coordination polyhedra with emphasized nitrogen atoms (circles) in the unit cell of 1. Projection onto the (101) plane. | ||
In both structures, π-π stacking of the aromatic rings occurs: the planes of the bpy or phen rings are oriented parallel, thus forming bpy-bpy and phen-phen pairs, respectively. The distance between the planes in any pair is approximately 3.5 Å. Two ligand molecules of a pair are centrosymmetrically oriented (Figs. 5 and 6). The packing of aromatic rings in 1 and 2 shows that there are two different orientations of the pairs. In the structure of 1 the [VO(O2)(ox)(bpy)]− complex anions are connected via hydrogen bonds formed between the oxygen atoms of complex anions and crystal water molecules as shown in Fig. 7.
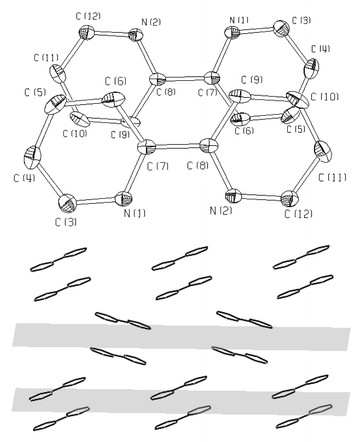 | ||
| Fig. 5 Detail of the π-π stacking: bpy-bpy pairs (top) and packing of the aromatic rings (bottom) along the 010 planes in the structure of 1. | ||
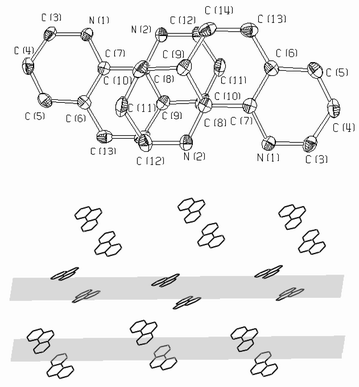 | ||
| Fig. 6 Detail of the π-π stacking: phen-phen pairs (top) and packing of the aromatic rings (bottom) along the 001 planes in the structure of 2. | ||
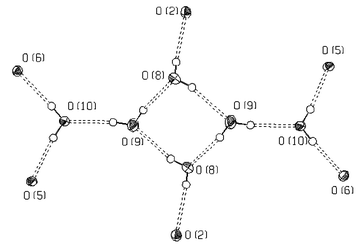 | ||
| Fig. 7 “Network unit” in the structure of 1 formed by hydrogen bonds between six water molecules (O8, O9, O10) and the O(2) peroxo and O(5), O(6) oxalato oxygen atoms originating from six complex anions. | ||
As follows from the data in Table 1, the anionic pic [pyridine-2-carboxylate(1−)]27 and ox ligands in the vanadium(V) monoperoxo complexes, with two different bidentate heteroligands, are preferably bound in two equatorial positions (EEP) while the neutral ligands bpy and phen bind in equatorial and apical positions (EAP). The small number of known structures of this type does not allow to generalize and rationalize this phenomenon. The steric properties of the heteroligand do not seem to be essential, because the four positions in the bis(heteroligand) complexes can both be occupied by two small or two voluminous ligands as well: [VO(O2)(bpy)2]+,28 [VO(O2)(phen)2]+,28 [VO(O2)(ox)2]3−,29,30 [VO(O2)(pca)2]−,31 [VO(O2)(pa)2]+32 and [VO(O2)(pic)2]−14 [where pca is pyrazine-2-carboxylate(1−) and pa is picolinamide].
| Complex | EEP (ligand X) | EAP (ligand Y) | Ref. |
|---|---|---|---|
|
a pic=pyridine-2-carboxylate(1−), ox=oxalate(2−), bpy=2,2-bipyridine, phen=1,10-phenanthroline.
|
|||
| [VO(O2)(pic)(bpy)] | pic | bpy | 27 |
| [VO(O2)(pic)(phen)] | pic | phen | 14 |
| [VO(O2)(ox)(bpy)]− | ox | bpy | This work |
| [VO(O2)(ox)(phen)]− | ox | phen | This work |
Raman and IR spectra
There is a very limited number of Raman spectral studies of vanadium monoperoxo complexes in solution and in the solid state.33,34 Based on the detailed 51V NMR and Raman spectral study of aqueous solutions of the peroxovanadates, Griffith et al.33 assigned the bands (with Raman intensities in parentheses) at 976 (8), 890 (4), 589 (2) and 542 (2) cm−1 to vibrations in the [VO(O2)(H2O)x]+ ion: ν(V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), ν(Op–Op)
(Op is the peroxo oxygen atom), νas[V(Op)2] and νs[V(Op)2], respectively. We consider this assignment to be very reliable, since the Raman bands corresponding to stretching vibrations of the V–O(H2O) groups are usually very weak.35 In agreement with this we assign the very strong Raman bands at 957 cm−1 for 1 and at 952 cm−1 for 2 to ν(VO) and the bands of medium intensity at 927 cm−1 for 1 and at 935 cm−1 for 2 to Op–Op stretching vibrations. These two bands, sometimes split due to crystal field effects (factor group splitting) and usually not affected by weaker bands of the heteroligands, are very characteristic for vanadium(V) monoperoxo complexes (Figs. 8 and 9). The bands corresponding to V–Op stretching vibrations at about 550 cm−1 could not be reliably distinguished from the bands arising from the vibrations of heteroligands in the same region.
O), ν(Op–Op)
(Op is the peroxo oxygen atom), νas[V(Op)2] and νs[V(Op)2], respectively. We consider this assignment to be very reliable, since the Raman bands corresponding to stretching vibrations of the V–O(H2O) groups are usually very weak.35 In agreement with this we assign the very strong Raman bands at 957 cm−1 for 1 and at 952 cm−1 for 2 to ν(VO) and the bands of medium intensity at 927 cm−1 for 1 and at 935 cm−1 for 2 to Op–Op stretching vibrations. These two bands, sometimes split due to crystal field effects (factor group splitting) and usually not affected by weaker bands of the heteroligands, are very characteristic for vanadium(V) monoperoxo complexes (Figs. 8 and 9). The bands corresponding to V–Op stretching vibrations at about 550 cm−1 could not be reliably distinguished from the bands arising from the vibrations of heteroligands in the same region.
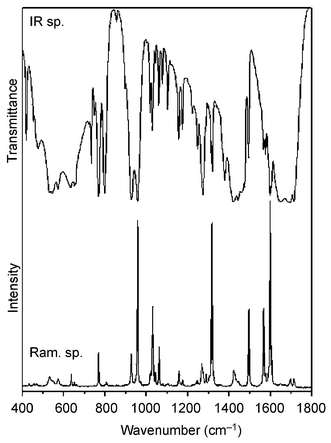 | ||
| Fig. 8 IR and Raman spectra of 1. | ||
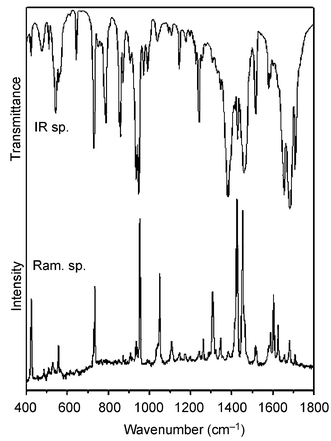 | ||
| Fig. 9 IR and Raman spectra of 2. | ||
The characteristic IR absorption bands (Figs. 8 and 9) corresponding to stretches in the VO(O2)+ moiety appear in the 1000–500 cm−1 region: ν(VO) at 957 cm−1 for 1 and 948 cm−1 for 2, ν(Op–Op) at 928 cm−1 for 1 and 935 cm−1 for 2, ν(V–Op) at 544 and 573 cm−1 for 1 and at 543 and 567 cm−1 for 2. Several bands corresponding to ν(COO) stretchings in the deprotonated carboxylic group occur at 1647–1713 cm−1 for 1 and at 1655–1709 cm−1 for 2. The other bands are in agreement with data published for the oxalate anion36,37 and for bpy (resp. phen).38–40
51V NMR spectroscopy
As seen from the 51V NMR spectra, the composition of water–ethanol and water–acetonitrile reaction solutions for the preparation of 1 (Fig. 10, spectrum I) and 2 (Fig. 11, spectrum I) is from the viewpoint of the number of species very simple when compared with the composition of some other systems, such as solutions of the peroxo vanadium species formed with imidazole or alanylhistidine.41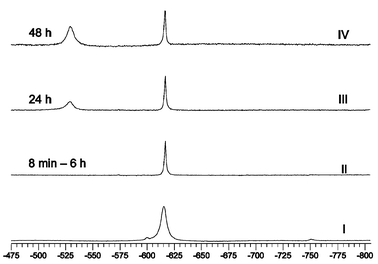 | ||
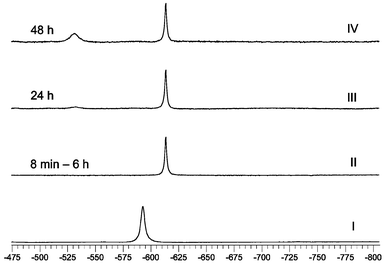
| Fig. 10
51V NMR spectra of 1. Spectrum I: reaction solution for the preparation of 1, conditions: 7 h after preparation, T=278 K, water–ethanol solvent, w(ethanol)=13%, pH 3.3, c(V)=c(H2ox)=c(bpy)=0.08 mol L−1, c(H2O2)=0.16 mol L−1. Spectra II–IV: aqueous solution of 1, 8 min to 48 h after dissolution, c=0.02 mol L−1, T=295 K. | ||
 | ||
| Fig. 11
51V NMR spectra of 2. Spectrum I: reaction solution for preparation of 2, conditions: 7 h after preparation, T=278 K, water–acetonitrile solvent, φ(acetonitrile)=53%, c(V)=c(H2ox)=c(phen)=c(Pr4NOH)=0.09 mol L−1, c(H2O2)=0.175 mol L−1. Spectra II–IV: aqueous solution of 2, 8 min to 48 h after dissolution, c=0.02 mol L−1, T=295 K. | ||
Spectra I of the reaction solutions (Figs. 10 and 11) exhibit the chemical shifts of the major species: [VO(O2)(ox)(bpy)]−
(δV=−615.4, Ir=92%) and [VO(O2)(ox)(phen)]−
(δV=−592.6, Ir=98.7%), as well as the shifts of minor species: [VO(O2)2(bpy)]−
(δV=−750.0, Ir=3.6%), [VO(O2)(ox)2]−
(δV=−600.1, Ir=4%) and [VO(O2)2(phen)]−
(δV=−731.8, Ir=1.3%).42 The difference in chemical shifts for the predominant species in the mixed solvent reaction solutions, when compared with pure aqueous solutions (spectra II–IV, Figs. 10 and 11), is caused by different temperature and solvation effects:43 the observed δV=−592.6 for [VO(O2)(ox)(phen)]− is between the δV values for 2 in acetonitrile (−555.7) and water (−612.9), respectively.
The spectra II of aqueous solutions of 1 and 2
(Figs. 10 and 11) exhibit a single signal at −615.5 ppm for 1 and at −612.9 ppm for 2, which allows one to suggest that the structure of both complex anions is maintained on dissolution. As concerns peroxo ligand decomposition, the complex anions differ in stability: 2 is more stable than 1 as the spectra of both complexes showed no decomposition within 6 h but after 24 h the Ir for 1 and 2 decreased to 52% (resp. 88%), and after 48 h to 32% (resp. 53%). Based on the data published in ref. 44 we assign the signals at −529.0 (resp. −531.2 ppm) to the peroxo free dioxo complexes formed on decomposition: [VO2(ox)(bpy)]−
{resp. [VO2(ox)(phen)]−). The slight asymmetry of the signals of [VO2(ox)(bpy)]− and [VO2(ox)(phen)]− can be explained by formation of the dioxo-oxalato complexes, [VO2(ox)2]3− and [VO2(ox)]−, with δV values of −536.2 and −532.6 ppm, respectively.44 We have also observed the same asymmetry in the spectrum of a dioxovanadium complex formed “in situ” in a vanadate–H2ox–bpy–HClO4–H2O solution [c(V)=c(ox)=c(bpy)=0.02 mol L−1, pH 3.68], which exhibits a peak at −530 ppm and a shoulder at −536 ppm. The “in situ” measurements with phen could not be performed due to the insolubility of phen in water. The 51V NMR measurements confirm that in the reaction solutions as well as in solutions of 1 and 2, only isomers with the same structure as that found by X-ray analysis are formed (Scheme 1, Path 1). The formation of isomers B and B′
via Path 2 or by isomerization of A and A′ was not observed.
![Formation and isomerization of [VO(O2)(ox)(bpy)]− and [VO(O2)(ox)(phen)]−.](/image/article/2004/NJ/b306090j/b306090j-s1.gif) | ||
| Scheme 1 Formation and isomerization of [VO(O2)(ox)(bpy)]− and [VO(O2)(ox)(phen)]−. | ||
Conclusions
New racemic vanadium(V) monoperoxo complexes, K[VO(O2)(ox)(bpy)]·3H2O and Pr4N[VO(O2)(ox)(phen)], were synthesized. The X-ray analysis revealed the typical distorted pentagonal bipyramidal geometry in both complex anions. The donor atoms of the bidentate anionic heteroligand ox occupy two equatorial positions of the pentagonal pseudo-plane, while the neutral bidentate ligands, bpy or phen, occupy one equatorial and one apical positions. Such an occupation of the coordination sites of the vanadium atom in the VO(O2) core in the presence of anionic and neutral bidentate ligands seems to be a general phenomenon; however, it must still be confirmed by structural data for a greater number of similar complexes. The 51V NMR spectra of aqueous reaction solutions and of complexes dissolved in water have confirmed that only the species with the same structure as that found in the solid state, but not the isomers based on ox↔bpy or ox↔phen ligand exchange, exist in solution.
Acknowledgements
This work was financially supported by the Ministry of Education of the Slovak Republic (Grant 1/8201/01) and by Comenius University, Bratislava (Grant 49/2001/UK). The authors wish to express their gratitude to Prof. Toužín from Masaryk University in Brno, Czech Republic, for the Raman spectral measurements.References
- H. Vilter, Bot. Mar., 1983, 26, 429 Search PubMed.
- A. Messerschmidt, L. Prade and R. Wever, Biol. Chem., 1997, 378, 309 CAS.
- K. H. Thompson and C. Orvig, Coord. Chem. Rev., 2001, 219–221, 1033 CrossRef CAS.
- K. Kanamori, K. Nishida, N. Miyata, K.-I. Okamoto, Y. Miyoshi, A. Tamura and H. Sakurai, J. Inorg. Biochem., 2001, 86, 649 CrossRef CAS.
- F. P. Ballistreri, E. G. M. Barbuzzi, G. A. Tomaselli and R. M. Toscano, J. Inorg. Biochem., 2000, 80, 173 CrossRef CAS.
- X. W. Zhou, Q.-X. Chen, Z. Chen, Z.-Q. He and H.-M. Zhou, Biochemistry (Moscow), 2000, 65, 1424 CrossRef CAS.
- H. Mimoun, L. Saussine, E. Daire, M. Postel, J. Fischer and R. Weiss, J. Am. Chem. Soc., 1983, 105, 3101 CrossRef CAS.
- N. N. Karpyshev, O. D. Yakovleva, E. P. Talsi, K. P. Bryliakov, O. V. Tolstikova and A. G. Tolstikov, J. Mol. Catal. A: Chem., 2000, 157, 91 CrossRef CAS.
- K. P. Bryliakov, N. N. Karpyshev, S. A. Fominsky, A. G. Tolstikov and E. P. Talsi, J. Mol. Catal. A: Chem., 2001, 171, 73 CrossRef CAS.
- C. Ohta, H. Shimizu, A. Kondo and T. Katsuki, Synlett, 2002, 161 CrossRef CAS.
- P. Schwendt, P. Švančárek, I. Smatanová and J. Marek, J. Inorg. Biochem., 2000, 80, 59 CrossRef CAS.
- F. W. B. Einstein, R. J. Batchelor, S. J. Angus-Dunne and A. S. Tracey, Inorg. Chem., 1996, 35, 1680 CrossRef CAS.
- P. Schwendt, A. Oravcová, J. Tyršelová and F. Pavelčík, Polyhedron, 1996, 15, 4507 CrossRef CAS.
- V. S. Sergienko, M. A. Porai-Koshits, V. K. Borzunov and A. B. Ilyukhin, Russ. J. Coord. Chem., 1993, 19, 714 Search PubMed.
- K. Kanamori, K. Nishida, N. Miyata, K.-I. Okamoto, Y. Miyoshi, A. Tamura and H. Sakurai, J. Inorg. Biochem., 2001, 86, 649 CrossRef CAS.
- C. Djordjevic, S. A. Craig and E. Sinn, Inorg. Chem., 1985, 24, 1281 CrossRef CAS.
- P. Švančárek, P. Schwendt, J. Tatiersky, I. Smatanová and J. Marek, Monatsh. Chem., 2000, 131, 145 CrossRef CAS.
- F. Demartin, M. Biagioli, L. Strinna-Erre, P. Angelo and G. Micera, Inorg. Chim. Acta, 2000, 299, 123 CrossRef CAS.
- P. Schwendt, P. Švančárek, L. Kuchta and J. Marek, Polyhedron, 1998, 17, 2161 CrossRef CAS.
- A. Messerschmidt and R. Wever, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 392 CrossRef CAS.
- S. Macedo-Ribeiro, W. Hemrika, R. Renirie, R. Wever and A. Messerschmidt, J. Biol. Inorg. Chem., 1999, 4, 209 CrossRef CAS.
- M. Sivák, D. Joniaková and P. Schwendt, Transition Met. Chem., 1993, 18, 304 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A, 1990, 46, 467 CrossRef.
- G. M. Sheldrick and T. R. Schneider, Methods Enzymol., 1997, 277, 319 CrossRef CAS.
- M. Nardelli, Comput. Chem., 1983, 7, 95 CrossRef CAS.
- (a) M. N. Burnett and C. K. Johnson, ORTEP-III: Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustrations, Report ORNL-6895, Oak Ridge National Laboratory, Oak Ridge, TN, USA, 1996 Search PubMed; (b) see http://www.chem.gla.ac.uk/~louis/software/struplo/.
- H. Szentivanyi and R. Stomberg, Acta Chem. Scand. Ser. A, 1982, 37, 709.
- V. S. Sergienko, V. K. Borzunov and M. A. Porai-Koshits, Zh. Neorg. Khim., 1992, 37, 1062 Search PubMed.
- R. Stomberg, Acta Chem. Scand. Ser. A, 1986, 40, 168.
- P. Schwendt, P. Švančárek, F. Pavelčík and J. Marek, Chem. Pap., 2002, 56, 158 Search PubMed.
- G. Süss-Fink, S. Stanislas, G. B. Shul'pin, G. V. Nizova, H. Stoeckli-Evans, A. Neels, C. Bobillier and S. Claude, J. Chem. Soc., Dalton Trans., 1999, 3169 RSC.
- M. Sivák, M. Mad'arová, J. Marek and J. Benko, Chem. Listy, Suppl., 2000, 94, 906 Search PubMed.
- N. J. Campbell, A. C. Dengel and W. P. Griffith, Polyhedron, 1989, 8, 1379 CrossRef CAS.
- P. Schwendt, M. Sivák, A. E. Lapshin, Y. I. Smolin, Y. F. Shepelev and D. Gyepesová, Transition Met. Chem., 1994, 19, 34 CAS.
- P. Schwendt and M. Pisárčik, Spectrochim. Acta, Part A, 1990, 46, 397 CrossRef.
- P. Schwendt, P. Petrovič and D. Úškert, Z. Anorg. Allg. Chem., 1980, 466, 232 CAS.
- J. Fujita, A. E. Martell and K. Nakamoto, J. Chem. Phys., 1962, 36, 324 CrossRef CAS.
- J. S. Strukl and J. L. Walter, Spectrochim. Acta, Part A, 1971, 27, 223 CrossRef CAS.
- J. S. Strukl and J. L. Walter, Spectrochim. Acta, Part A, 1971, 27, 209 CrossRef CAS.
- D. A. Thornton and G. M. Watkins, J. Coord. Chem., 1992, 25, 299 CAS.
- L. Pettersson, I. Andersson and A. Gorzsás, Coord. Chem. Rev., 2003, 237, 77 CrossRef CAS.
- C. Weidemann, W. Priebsch and D. Rehder, Chem. Ber., 1989, 122, 235 CrossRef CAS.
- V. Conte, F. Di Furia and S. Moro, Inorg. Chim. Acta, 1998, 272, 62 CrossRef CAS.
- P. M. Ehde, L. Pettersson and J. Glaser, Acta Chem. Scand., 1991, 45, 998 CAS.
Footnote |
| † CCDC reference numbers 212922 and 212923. See http://www.rsc.org/suppdata/nj/b3/b306090j/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2004 |