Modeling the catalyst resting state in aryl tin(IV) polymerizations of lactide and estimating the relative rates of transamidation, transesterification and chain transfer†
Malcolm H.
Chisholm
*,
Ewan E.
Delbridge‡
and
Judith C.
Gallucci
Department of Chemistry, The Ohio State University, 100 W 18th Avenue,, Columbus,, OH 43210-1185, U.S.A.. E-mail: chisholm@chemistry.ohio-state.edu; Fax: +1 614 292 0368; Tel: +1 614 292 7216
First published on 21st November 2003
Abstract
The preparation and characterization (IR, 1H, 13C{1H}, 119Sn NMR spectroscopy, elemental analysis and single crystal X-ray structure determination) are reported for Ph3SnOCMe2C(O)OEt (1) and Ph2Sn[OCMe2C(O)NMe2]2 (2). In the solid state, compound 1 contains four-coordinate tin with evidence for incipient bond formation to the ester oxygen: Sn⋯O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =2.648(2)
Å. Compound 2 contains six-coordinate tin in a pseudo-octahedral geometry. The OCMe2C(O)NMe2 groups form cis-chelates with short, ca. 2.03 Å, and long, ca. 2.26 Å, Sn–O bonds to alkoxide and amide oxygen atoms, respectively. In solution, compound 1 remains four-coordinate but compound 2 exists as an equilibrium mixture of six-coordinate and five-coordinate species as judged by NMR spectroscopy. At −50°C in toluene-d8, the six-coordinate isomer is favored and the NMR data are consistent with the structure observed in the solid state. At +50°C, the NMR data are consistent with a five-coordinate species in which reversible chelation of η2- and η1-OCMe2C(O)NMe2 is fast on the NMR time scale. The molecular structure of 2 and its dynamic solution behavior is proposed to resemble that of Ph2Sn[OCHMeC(O)NMe2]2 formed in the polymerization of L-lactide by Ph2Sn(NMe2)2. The high formation tendency of this compound is proposed to be responsible for the preferential formation of cyclic lactide oligomers (LA/2)n by intrachain transesterification, in contrast to polymerizations employing Ph2Sn(OPri)2, which produce long chains of H–(LA/2)n–OPri where LA=[OCHMeC(O)OCHMeC(O)]. The kinetics of the reactions between Ph3SnX and each of Me2CHC(O)OMe, Me(MeO)CHC(O)OEt and Ph3SnOCHMeC(O)OEt, have been determined from NMR studies in benzene-d6 where X=NMe2 or OPri. Similarly, the reaction between Ph3SnOBut and (p-tolyl)3SnOPri has been followed. The former reactions represent transamidation and transesterification, and the latter models chain transfer. These findings, when compared to the earlier studies of the ring-opening of lactide and its subsequent ring-opening polymerization, indicate that the rate follows the order: chain transfer>ring-opening>ring-opening polymerization>transesterification, although the latter is influenced by the ester end-group.
=2.648(2)
Å. Compound 2 contains six-coordinate tin in a pseudo-octahedral geometry. The OCMe2C(O)NMe2 groups form cis-chelates with short, ca. 2.03 Å, and long, ca. 2.26 Å, Sn–O bonds to alkoxide and amide oxygen atoms, respectively. In solution, compound 1 remains four-coordinate but compound 2 exists as an equilibrium mixture of six-coordinate and five-coordinate species as judged by NMR spectroscopy. At −50°C in toluene-d8, the six-coordinate isomer is favored and the NMR data are consistent with the structure observed in the solid state. At +50°C, the NMR data are consistent with a five-coordinate species in which reversible chelation of η2- and η1-OCMe2C(O)NMe2 is fast on the NMR time scale. The molecular structure of 2 and its dynamic solution behavior is proposed to resemble that of Ph2Sn[OCHMeC(O)NMe2]2 formed in the polymerization of L-lactide by Ph2Sn(NMe2)2. The high formation tendency of this compound is proposed to be responsible for the preferential formation of cyclic lactide oligomers (LA/2)n by intrachain transesterification, in contrast to polymerizations employing Ph2Sn(OPri)2, which produce long chains of H–(LA/2)n–OPri where LA=[OCHMeC(O)OCHMeC(O)]. The kinetics of the reactions between Ph3SnX and each of Me2CHC(O)OMe, Me(MeO)CHC(O)OEt and Ph3SnOCHMeC(O)OEt, have been determined from NMR studies in benzene-d6 where X=NMe2 or OPri. Similarly, the reaction between Ph3SnOBut and (p-tolyl)3SnOPri has been followed. The former reactions represent transamidation and transesterification, and the latter models chain transfer. These findings, when compared to the earlier studies of the ring-opening of lactide and its subsequent ring-opening polymerization, indicate that the rate follows the order: chain transfer>ring-opening>ring-opening polymerization>transesterification, although the latter is influenced by the ester end-group.
Introduction
Biodegradable and biocompatible polymers formed from inexpensive renewable resources are attracting considerable current attention in both academic and industrial circles.1–3 For example, polylactides (PLAs) formed by the ring-opening polymerization (ROP) of lactide (LA), find numerous applications ranging from environmentally friendly bulk packaging materials4 to control-released drug delivery agents1,5, artificial sutures6 and polymer matrices for tissue engineering.1,7 Cargill–Dow has entered a joint venture for the production of PLA on the order of 3×108 1b per annum based on an enzymatic process for the formation of LA from corn. Ring-opening polymerization can be brought about in a melt with catalysts such as tin(II) octanoate, or in a more controlled manner in solution by a variety of well-defined coordination catalyst precursors3 or organic bases in the presence of alcohol initiators.8 An excellent recent review documents work employing coordination catalysts.3 Work by Ovitt and Coates2i has recently shown that stereochemical control in the polymerization of meso-lactide can generate syndiotactic-PLA and rac-LA can be converted to heterotactic-PLA by what appears to be end-group control of the ring-opening event at sterically demanding zinc metal centers. Control of polymer microstructure as well as molecular weight and molecular weight distribution, together with the preparation of co-polymers and polymer blends involving LA, represent important challenges in what is emerging as a significant new field in polymer science.
In the ring-opening polymerization of LA by a coordination metal complex, the active species contains a metal–alkoxide bond. This metal–oxygen bond is kinetically labile to other reactions, namely chain transfer and transesterification. An earlier claim9 that rac-salen Al–OR complexes could polymerize rac-LA to the stereoplex polymer [poly(L-LA)+poly(D-LA)] was refuted2i,j based on an analysis of the mistakes in the polymer ,which revealed that the polymer was a blocky polymer of (L-LA)n–(D-LA)m where n∼m∼10 or 11. Chain transfer in conjunction with ring-opening polymerization would give such a polymer. The addition of an alcohol would also lead to chain transfer and limit molecular weight by increasing the number of growing chains. The process of transesterification leads to loss of stereochemistry in the resulting polymer and also leads to a broadening of the molecular weight distribution with time. It is, however, a process by which LA can be introduced into other polyesters, thus providing a potential route to new co-polymers and blends.
In our studies of ring-opening polymerization of lactide by aryl tin(IV) complexes, we observed both chain transfer and transesterification.10 Furthermore, when Sn–NMe2 groups were used as initiators, we observed a marked preference for the formation of cyclic oligomers of PLA, (LA/2)n, along with long chains of H(LA/2)nNMe2 [where LA=OCHMeC(O)OCHMeC(O)], when compared with Sn–OPri initiators. Since these tin(IV) catalyst systems are relatively slow, we were determined to examine the origin of these effects and to try to understand the fundamental reactions involved. We describe here our findings, which were prompted by these considerations.
Results and discussion
Syntheses
The synthesis of aryl tin alkoxides and dimethylamides has been described previously.10 The details of the new tin alkoxyester synthesis are described in the experimental. The formation of Ph2Sn[OCMe2C(O)NMe2]2, compound 2, in the reaction between Ph2Sn(NMe2)2 and HOCMe2C(O)OEt is not immediately obvious. However, it can be reasonably well understood in terms of the reaction sequence displayed in Scheme 1. Following the initial alcoholysis there is a rapid intramolecular amidation reaction leading to formation of Ph2Sn(OEt)[OCMe2C(O)NMe2)], which then undergoes ligand (alkoxide) exchange to form the stable bis chelate complex 2.![Pathway to the formation of Ph2Sn[OCMe2C(O)NMe2]2 (2).](/image/article/2004/NJ/b306700a/b306700a-s1.gif) | ||
| Scheme 1 Pathway to the formation of Ph2Sn[OCMe2C(O)NMe2]2 (2). | ||
An attempt was made to prepare the ester analog of 2, namely Ph2Sn[OCMe2C(O)OEt]2, from the reaction between Ph2SnCl2 and two equivalents of LiOCMe2C(O)OEt. However, the product of this reaction was very insoluble in organic solvents, which suggests it may be polymeric with µ-OR bridges. That the compound is not similar to 2 serves to demonstrate the significant difference in the chelating ability of the OCMe2COX ligands where X=NMe2 and OEt, though ethyl lactate has been seen to chelate zinc(II) in the solid state.2g
Solid state and molecular structures
A summary of crystal data for compounds Ph3SnOCMe2C(O)OEt (1) and Ph2Sn[OCMe2C(O)NMe2]2 (2) is given in Table 1 and ORTEP drawings of the molecular structures are displayed in Figs. 1 and 2, respectively. Selected bond lengths and angles are given in Tables 2 and 3. Compound 1 can be considered to contain a four-coordinate Sn(IV) center with evidence of incipient Sn⋯O bond formation (Sn⋯O=2.65 Å) to the ketonic carbon of the ester. However, the molecule clearly does not contain a five-coordinate Sn(IV) center as seen, for example, in the tropinato complex Ph3Sn(trop)11
(shown in Fig. 3). In contrast, compound 2 most definitely contains six-coordinate Sn(IV). The Sn–O bonds fall into two groups, short bonds with Sn–O ca. 2.03 Å to the alkoxide oxygen atoms and long bonds with Sn–O ca. 2.26 Å to the amide oxygen atoms. Again, a comparison can be made with the tropanato complex Me2Sn(trop)211
(shown in Fig. 3). As indicated below the comparison is pertinent not only from a structural standpoint but also in terms of the solution NMR spectroscopy of these molecules.
| 1 | 2 | |
|---|---|---|
|
a
R
1=Σ||Fo|−|Fc||/Σ|Fo|.
b
wR
2=[Σw(F2o−F2c)2/Σw(F2o)2]1/2.
|
||
| Formula | C24H26O3Sn | C24H34N2O4Sn |
| FW | 481.14 | 533.22 |
| Crystal system | Triclinic | Monoclinic |
| Wavelength/Å | 0.71073 | 0.71073 |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
| a/Å | 9.433(1) | 11.554(1) |
| b/Å | 10.255(1) | 11.075(1) |
| c/Å | 12.180(2) | 19.704(2) |
| α/° | 81.66(1) | 90 |
| β/° | 87.44(1) | 98.947(4) |
| γ/° | 73.95(1) | 90 |
| U/Å3 | 1120.3(2) | 2490.8(4) |
| Z | 2 | 4 |
| T/K | 200(2) | 200(2) |
| Total reflections | 30902 | 44002 |
| Independent reflections | 5108 | 5704 |
| R int | 0.030 | 0.030 |
| Obs. reflections [I>2σ(I)] |
4503 | 4678 |
|
R
1
[I>2σ(I)]a |
0.024 | 0.0349 |
|
wR
2
[I>2σ(I)]b |
0.0527 | 0.0926 |
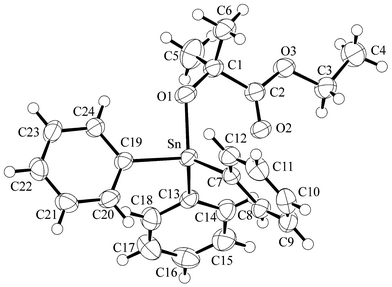 | ||
| Fig. 1 ORTEP plot of 1 showing 50% probability ellipsoids. | ||
![ORTEP plot of 2 showing 40% probability displacement ellipsoids. Hydrogen atoms omitted for clarity. Only one orientation of the disordered phenyl ring [at C(19)] is shown.](/image/article/2004/NJ/b306700a/b306700a-f2.gif) | ||
| Fig. 2 ORTEP plot of 2 showing 40% probability displacement ellipsoids. Hydrogen atoms omitted for clarity. Only one orientation of the disordered phenyl ring [at C(19)] is shown. | ||
| Sn–O(1) | 1.996(1) | O(1)–Sn–C(7) | 113.25(7) |
| Sn–C(7) | 2.126(2) | O(1)–Sn–C(13) | 118.73(7) |
| Sn–C(13) | 2.141(2) | O(1)–Sn–C(19) | 93.86(6) |
| Sn–C(19) | 2.148(2) | C(7)–Sn–C(13) | 114.44(7) |
| Sn⋯O(2) | 2.648(2) | C(7)–Sn–C(19) | 108.47(7) |
| C(13)–Sn–C(19) | 105.05(7) |
| Sn–O(1) | 2.267(2) | Sn–O(4) | 2.024(2) |
| Sn–O(2) | 2.037(2) | Sn–C(13) | 2.149(3) |
| Sn–O(3) | 2.258(2) | Sn–C(19) | 2.146(3) |
| O(1)–Sn–O(2) | 74.02(8) | O(2)–Sn–C(19) | 102.97(13) |
| O(1)–Sn–O(3) | 77.18(8) | O(3)–Sn–O(4) | 73.99(8) |
| O(1)–Sn–O(4) | 86.59(9) | O(3)–Sn–C(13) | 89.97(9) |
| O(1)–Sn–C(13) | 162.09(10) | O(3)–Sn–C(19) | 163.84(12) |
| O(1)–Sn–C(19) | 91.61(11) | O(4)–Sn–C(13) | 101.96(11) |
| O(2)–Sn–O(3) | 85.29(8) | O(4)–Sn–C(19) | 93.99(13) |
| O(2)–Sn–O(4) | 154.38(9) | C(13)–Sn–C(19) | 103.30(12) |
| O(2)–Sn–C(13) | 92.76(11) |
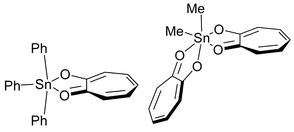 | ||
| Fig. 3 Molecular structures of Ph3Sn(trop) and Me2Sn(trop)2. | ||
NMR spectral properties of compounds 1 and 2
Compound 1 displays NMR behavior in benzene-d6 typical of a four-coordinate Ph3SnOR compound. In particular, the 119Sn resonance is at δ −143, which contrasts with five-coordinate Sn(IV) signals that are found around δ −200. That of Ph3Sn(trop) is reported at δ −182,11 for example. In contrast, compound 2 shows interesting temperature-dependent NMR spectra (Fig. 4 and 5), which is uncommon for R2SnX2 compounds. At low temperatures the 1H NMR spectra are consistent with the structure seen in the solid state (Fig. 5). Notably there are two NMe singlets, consistent with restricted rotation about the C–N partial double bond due to the resonance structure shown in Fig. 6. The CMe2 methyl groups are diastereotopic as a result of the virtual C2 symmetry of the molecule. At low temperatures (−50°C and below) there is just one 119Sn resonance at δ
−383, which predominates, and which may be compared with the Ph2Sn(trop)2 compound for which a peak at δ
−359 is reported.11
![119Sn NMR of Ph2Sn[OCMe2C(O)NMe2]2 (2) performed at high and low temperatures in toluene-d8.](/image/article/2004/NJ/b306700a/b306700a-f4.gif) | ||
| Fig. 4 119Sn NMR of Ph2Sn[OCMe2C(O)NMe2]2 (2) performed at high and low temperatures in toluene-d8. | ||
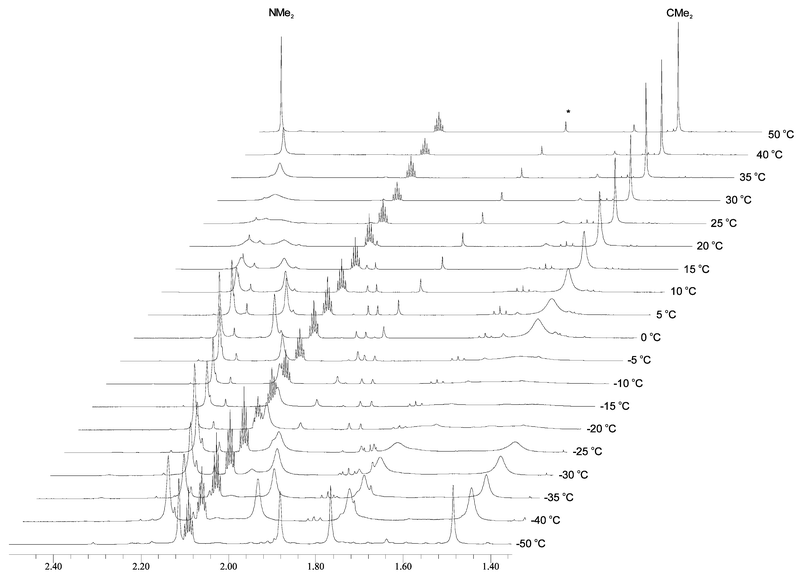 | ||
| Fig. 5 Variable temperature 1H NMR spectra of 2 measured in toluene-d8 showing the methyl region. * denotes a small impurity and the pentet is due to toluene-d8. | ||
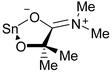 | ||
| Fig. 6 Resonance structure of the amide. | ||
Upon raising the temperature the 1H signals associated with the two CMe2 and two NMe2 resonances broaden, coalesce and above 50°C appear as sharp signals (Fig. 5). This is consistent with dechelation to give a five- or four-coordinate Sn(IV) centers. A five-coordinate monochelate of the structural type shown in Fig. 7 would have a mirror plane of symmetry and with rapid and reversible chelation would lead to a single CMe2 proton signal. With dechelation, rotation about the C–N bond of the amide group would allow N-methyl site exchange, as is well-known for organic amides such as DMF.
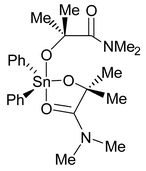 | ||
| Fig. 7 Five-coordinate representation of 2. | ||
The variable temperature 119Sn NMR spectra (Fig. 4) are particularly informative and support the view that at room temperature and above the predominant species present in solution is a five-coordinate Sn(IV) complex with δ −276 (Fig. 4). Paricularly pertinent to the discussion that follows is the fact that similar NMR behavior was observed for Ph2Sn[OCHMeC(O)NMe2]2, which is the compound formed in the reaction between Ph2Sn(NMe2)2 and L-lactide (1 equiv.); it is also the resting state of the tin catalyst in the ring-opening polymerization (ROP) reaction between Ph2Sn(NMe2)2 and excess L-lactide when the L-lactide has been consumed.10 Furthermore, Ph2Sn[OCHMeC(O)NMe2]2is formed in the reaction between Ph3SnNMe2 and L-lactide by disproportionation of Ph3SnOCHMeC(O)NMe2 into Ph4Sn and Ph2Sn[OCHMeC(O)NMe2]2.10 The compound Ph3SnOCHMeC(O)OCHMeC(O)NMe2 also is kinetically labile to the formation of Ph3SnOCHMeC(O)NMe2 and long chain Ph3SnOCHMeC(O)–(LA/2)n–OCHMeC(O)NMe2 by intermolecular transesterification. It is the thermodynamic preference for chelation of the OCHMeC(O)NMe2 ligand to the Sn(IV) center, as seen in the structure of the model compound 2, and in its solution behavior that is dominating the behavior of the Ph2Sn(NMe2)2 catalyst systems in the ROP of L-lactide.
Transesterification and transamidation reactions
A series of bimolecular reactions was investigated as outlined in Scheme 2. These reactions attempt to model transesterification, transamidation and chain transfer reactions, which are prevalent in the ROP of lactide. Both the steric and electronic effects of the substrates have been considered and how they influence the rates of product formation. It is apparent from Scheme 2 that the transamidation reactions A–C are not equilibrium reactions but are quantitative (>95% conversion) since a tin–amide bond is activated and forms the thermodynamically more stable tin–alkoxide bond. Conversely, in the transesterification reactions D and E a tin–alkoxide bond is activated to form another tin–alkoxide, bond resulting in an equilibrium being established where at t∞ all four species, [A], [B], [C] and [D], are in equal concentration (see Experimental). It is also important to note that none of the transesterification reactions occur at room temperature whilst the transamidation reactions do. Kinetic data were ascertained from 1H NMR data collected on samples made up in benzene-d6 (see Experimental and Electronic supplementary information for details).![Transamidation, transesterification and chain transfer reactions between tin(iv) compounds and organic moieties [T = 26(1) °C].](/image/article/2004/NJ/b306700a/b306700a-s2.gif) | ||
| Scheme 2 Transamidation, transesterification and chain transfer reactions between tin(IV) compounds and organic moieties [T=26(1)°C]. | ||
=4.7(2)×10−4 M−1 s−1] than the free esters [reactions A and B; k=7.8(2)×10−4 to 1.4(2)×10−2 M−1 s−1]. This unexpected result is in contrast with that found for transesterification reactions where reaction D proceeds more rapidly than reaction E. Based on electronic arguments alone reaction C would be expected to proceed more rapidly than reaction A(i), which would be consistent with the relative rates seen below for transesterification reactions D and E. A plausible explanation for this apparent contradiction in relative rates in these transamidation and transesterification reactions may lie in the fact that steric hindrance of Ph3Sn maybe more dominant in reaction C than in reaction D. Reaction C is performed at room temperature where the ketonic chelation to tin is more dominant than in reaction D, which is carried out at 60°C. Under these lower temperature conditions the bulky Ph3Sn group would more effectively hinder access to the ketonic carbon, resulting in suppression of reaction rate for the room temperature reaction. It is interesting to compare reactions rates for reactions A(i) and B [k=7.8(2)×10−4vs. 1.4(2)×10−2 M−1 s−1] where replacing one methyl with a methoxy group on the ester leads to a faster rate of reaction. This may be expected if the methoxy group is considered inductively electron withdrawing, as this would increase the electrophilicity of the carbonyl carbon and thus its susceptibility to nucleophilic attack by NMe2. We also investigated the effect of adding an inert organic amide such as DMF (5 equiv.) to the reaction mixture [reactions A(i) and (ii)] and found that it suppressed the reaction rate [k=7.8(2)×10−4 to 5.2(2)×10−4 M−1 s−1]. This can be attributed to DMF competing with Me2CHC(O)OMe for coordination to the Ph3SnNMe2 prior to nucleophilic attack by NMe2 on the ester. Coordination is presumably a fast equilibrium process, forming an adduct that can then undergo reaction as shown below: =26(1)°C]. It is interesting to compare reactions D [Ph3SnOCHMeC(O)OEt+Ph3SnOPri; k=2.8(2)×10−3 M−1 s−1] and E [MeOCHMeC(O)OEt+Ph3SnOPri; k=4.0×10−5 M−1 s−1] in Scheme 2 where the rates of reaction are in the reverse order of that found in the corresponding transamidation but are consistent with the electron-withdrawing effect of the Ph3Sn group (see above for discussion).
=26(1)°C]. It is interesting to compare reactions D [Ph3SnOCHMeC(O)OEt+Ph3SnOPri; k=2.8(2)×10−3 M−1 s−1] and E [MeOCHMeC(O)OEt+Ph3SnOPri; k=4.0×10−5 M−1 s−1] in Scheme 2 where the rates of reaction are in the reverse order of that found in the corresponding transamidation but are consistent with the electron-withdrawing effect of the Ph3Sn group (see above for discussion).
As observed for the transamidation reactions the electronic properties of the ester affect its reactivity towards transesterification with Ph3SnOPri such that no reaction is observed in reaction F [Me2CHC(O)OMe+Ph3SnOPri] at 60°C. The electron-withdrawing properties of the MeO group in the ester in reaction E enhances reactivity such that transesterification can occur, albeit slowly.
It is also clear that steric factors play a significant role in transesterification reactions and this is illustrated in the comparison between reactions D and G where the –OCHMeC– backbone has been replaced by –OCMe2C–. The substitution of H for a second methyl group has provided sufficient protection such that no discernible reaction was observed at 60°C in reaction G. This is not surprising since we have previously observed no transesterification for Ph3SnOCMe2C(O)OCHMeC(O)NMe2.
We have also explored the steric effect of the nucleophile (OR) and its ability to undergo transesterification with the ester group in Ph3SnOCHMeC(O)OCHMeC(O)NMe2
(reaction H). It was found that reaction of Ph3SnOPri with Ph3SnOCHMeC(O)OCHMeC(O)NMe2 yielded two major products, Ph3SnOCHMeC(O)X where X=NMe2 or OPri; the OPri product results from transesterification with Ph3SnOPri. However, the corresponding reaction with Ph3SnOBut containing the bulkier OBut nucleophile only yielded one major product, namely Ph3SnOCHMeC(O)NMe2. This product, which is present in both reactions in H, results from Ph3SnOCHMeC(O)OCHMeC(O)NMe2 molecules reacting together in an intermolecular self-transesterification reaction to form Ph3SnOCHMeC(O)NMe2 and Ph3Sn[OCHMeC(O)]1.5NMe2. Further transesterification leads to Ph3SnOCHMeC(O)NMe2 and long chains of Ph3Sn–(LA/2)n–OCHMeC(O)NMe2, together with some cycles of PLA.
To summarize, these transesterification reactions are sensitive to not only the electronic and steric environments around the ester group but also those of the nucleophile. The transamidation reactions are also sensitive to the electronic environment of the ester group.
Chain transfer reactions
Chain transfer reactions involving the propagating lactide chain are important as polymer molecular weight can be conveniently modified via addition of a transfer agent such as an alcohol. As such, we wanted to quantify the ability of simple alkoxides, which mimic a propagating polylactide chain, to exchange between triaryl tin(IV) centers. Simple alkoxides were chosen instead of more complicated OR ligands that more closely resemble a lactide chain since simple triaryl tin(IV) alkoxides are not susceptible to transesterification reactions or indeed aryl group migration, as seen for Ph3Sn[OCHMeC(O)]nX compounds. At room temperature the reaction of Ph3SnOBut and (o-MeC6H4)3SnOPri in benzene-d6 proceeded rapidly towards equilibrium [k=3.0(2)×10−3 M−1 s−1; see reaction I in Scheme 2]. It is important to note that every attempt was made to remove even trace amounts of free alcohol from these reagents since free alcohol was found to drastically increase the rate of the alkoxide transfer. Moisture was rigorously excluded from the reagents and solvents and the reagents were heated under vacuum for an extended period to remove trace alcohol prior to their use in this reaction.
Kinetic summary
From this and our previous two studies,10 it is now possible to estimate the relative rates of the various processes in the ROP of lactide, namely ring-opening, propagation, transesterification and chain transfer. This allows us to order or rank the various rates of these processes (Table 4) such that the rate of chain transfer (kct)>transesterification (ktrans)>ring-opening (kro)>propagation (kprop). Note that the order is slightly different if one considers the metal-containing transesterification reaction with Ph3SnOCHMeC(O)OEt rather than the organic ester MeOCHMeC(O)OEt, where for the organic ester the order would switch to kct>kro>kprop>ktrans. This indicates that transesterification near a metal center is much more facile than somewhere along the polymer chain.
| A | B | T/°C | k/M−1 s−1 |
|---|---|---|---|
| a This work. b 1∶100 [Sn]∶[L-lacitide]. c Pseudo 1st order (s−1). d Previous work. | |||
| Ph3SnOPri | L-Lactide | 61 |
k
ro=1.3(2)×10−4a |
| Ph2Sn(OPri)2 | L-Lactideb | 61 |
k
prop=2.6(2)×10−5 s−1cd |
| Ph3SnOPri | MeOCHMeC(O)OEt | 60 |
k
trans=4.0(2)×10−5a |
| Ph3SnOPri | Ph3SnOCHMeC(O)OEt | 60 |
k
trans=2.8(2)×10−3a |
| Ph3SnOBut | (o-MeC6H4)3SnOPri | 26 |
k
ct=3.0(2)×10−3a |
Conclusions
This study of kinetically slow reactions of Sn(IV) compounds has given considerable insight into the intricacies of the polymerization of lactide by Ph2SnX2 and Ph3SnX compounds and its competing side reactions. We have quantified the rates of chain transfer, transesterification and transamidation reactions and compared some of these with previously determined ring-opening and propagation rates and found that the rates of such reactions follow kct>kro>kprop>ktrans or kct>ktrans>kro>kprop, depending on which transesterification reaction is employed. This is because transesterification occurs more rapidly near a metal center than in the absence of one (such as in the middle of a polymer chain). It should be noted that whilst we feel this overall order of reactivity to be accurate, these rate constants are derived from different reactions. In spite of this, and some similarities in some of the rate constant values, it is still possible to draw meaningful conclusions about the relative rates of these processes, which are prevalent in ROP lactide reactions. Overall it was found that transamidation reactions are in general faster than transesterification reactions but both are slower than chain transfer reactions.
This study has also investigated some solid state and solution properties of two key tin(IV) compounds. Both the coordination number and structure of Ph3SnOCMe2C(O)OEt (1) and Ph2Sn[OCMe2C(O)NMe2]2 (2) have been determined and the ketonic oxygen exhibits much stronger coordination to tin in Ph2Sn[OCMe2C(O)NMe2]2 than in Ph3SnOCMe2C(O)OEt. This is an important finding since previously we have found that the ability of the ketonic group to coordinate to the metal center influences many properties of polylactide formation, including molecular weight distribution and the ratio of polymer cycles to chains.
Experimental
General methods and materials
Caution: organotin(IV) compounds are highly toxic and require appropriate handling!The manipulation of air-sensitive compounds involved standard Schlenk line and dry box techniques. All solvents were distilled under nitrogen from alkali metals (sodium or sodium/potassium alloy) and stored over 4 Å molecular sieves. Benzene-d6, toluene-d8, ethyl L-(−)-lactate and ethyl (L)-(−)-2-methoxypropionate were purchased from Acros Scientific while ethyl 2-hydroxyisobutyrate was purchased from Aldrich and diphenyltin(IV) dichloride was purchased from Alfa Aesar. Tetrahydrofuran-d8 was purchased from Cambridge isotopes. Ethyl (L)-(−)-2-methoxypropionate was purified by column chromatography (diethyl ether–pentane) prior to drying and degassing. All esters were degassed and dried over activated molecular sieves. Benzene-d6, tetrahydrofuran-d8 and toluene-d8 were dried over sodium and vacuum transferred to a Schlenk flask containing activated molecular sieves. Ph3SnX, (o-MeC6H4)3SnOPri and Ph2SnX2 [where X=NMe2, OR or OCHMeC(O)OCHMeC(O)NMe2] were synthesized as described previously.10
1H NMR spectra were obtained from either Bruker DPX-400 or DRX-500 NMR spectrometers using either benzene-d6, toluene-d8 or tetrahydrofuran-d8. Spectra were referenced internally to the residual protio impurities for 1H (benzene-d6δ 7.15; toluene-d8δ 2.09; tetrahydrofuran-d8δ 1.73) and 13C (benzene-d6δ 128) or externally to Me4Sn for 119Sn (δ 0.0). Infrared data were obtained from a Perkin–Elmer Spectrum GX spectrophotometer with samples sandwiched between potassium bromide or sodium chloride plates as Nujol mulls (solids) or as neat liquids/semi-solids. Mass spectra were obtained from a Mircomass QTOF mass spectrometer. Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, GA on samples sealed under an inert atmosphere in glass ampoules.
Reaction kinetics
All kinetic data were obtained from NMR scale reactions. Standard solutions of Ar3SnX (X=NMe2, OPri, OBut; Ar=Ph, o-MeC6H4) and the appropriate ester [or tin(IV) containing compound] were made in benzene-d6 and stored in the dry box. Appropriate aliquots 1∶1 of both reagents were transferred to a J. Young® NMR tube. The total volume was made up to 800 µL with benzene-d6 to ensure a constant initial Ar3SnX concentration (0.0388 M). The reaction temperatures were regulated via a thermostatically controlled oil bath. Room temperature reactions were performed in air at 25°C.
An overall second-order process, first-order in both Ph3SnX [A] and ester or Sn(IV) containing compound [B], was assumed. Two different rate laws were used depending on whether the process was an equilibrium or not, that is A+B→C+D or A+B↔C+D. In either case the disappearance of both [A] and/or [B] was determined based on the formation of [C] and [D]. For non-equilibria cases plotting ln([B]/[A])
vs. time(s) produced a straight line {initial concentrations of A were 0.0388 M (for Ph3SnNMe2) and of B were 0.0310 [Ph3SnOCHMeC(O)OEt], 0.0335 [MeOCHMeC(O)OEt] and 0.0327 M [Me2CHC(O)OMe]}. The rate constants for these reactions were determined from the gradient of the graph viam=k([Bo]−[Ao]). In the equilibria cases, the derived rate law of King12 was used:
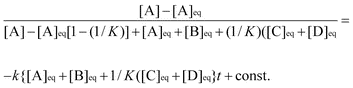
Syntheses
°C 10 mL) of dimethylamidotriphenyltin(IV)
(0.39 g, 1.00 mmol) ethyl L-(−)-lactate (125 µL, 1.10 mmol) was added slowly. The colorless reaction was stirred for 30 min after which time the hexane was removed, giving 0.42 g (90%) of an opaque semi-solid. Anal. calcd for C23H24O3Sn: C, 59.14; H, 5.18; found: C, 58.56; H, 4.56. IR (liquid): ν/cm−1 3065 m, 3050 m, 2980 m, 1710 br vs, 1481 m, 1447 w, 1439 s, 1375 m, 1335 w, 1302 m, 1235 br s, 1150 br s, 1076 s, 1057 m, 1023 m, 997 m, 941 w, 859 w, 799 vw, 729 s, 698 s, 659 m, 540 w, 492 w, 450 w.1H NMR (500 MHz, benzene-d6): δ 0.68 (t, OCH2Me, 3H), 1.40 (d, SnOCHMe, 3H), 3.64 (q, OCH2Me, 2H), 4.64 [q, SnOCHMe, 3H, 119/117Sn satellites JSnH (117Sn) 54, (119Sn) 40 Hz], 7.23 (t, m-H, 6H), 7.17 (d, p-H, 3H), 7.91 ([dd, o-H, 6H, JHH 4.6 and 1.9 Hz, 119/117Sn satellites JSnH (117Sn) 64, (119Sn) 48 Hz].13C{1H} NMR (126 MHz, benzene-d6): δ 13.81 (s, OCH2Me), 22.99 (s, SnOCHMe), 61.76 (s, OCH2Me), 68.99 (s, SnOCH2Me, 119/117Sn satellites JSnC 34 Hz), 128.72 (s, m-C, 119/117Sn satellites JSnC 60 Hz), 129.49 (s, p-C 119/117Sn satellites JSnC 13 Hz), 137.31 (s, o-C, 119/117Sn satellites JSnC 57 Hz), 142.52 (s, ipso-C), 181.13 [s, SnOCH(Me)C(O)]. 119Sn NMR (187 MHz, benzene-d6): δ
−129 (s). ESI-HR-MS: m/z calcd for C23H24O3Sn (MNa+): 491.0650; found: 491.0623 (5.5 ppm).
°C 10 mL) of dimethylamidotriphenyltin(IV)
(0.39 g, 1.00 mmol) ethyl 2-hydroxyisobutyrate (150 µL, 1.10 mmol) was added slowly. The colorless reaction was stirred for 30 min after which time the hexane was removed, giving 0.46 g (96%) of a white solid. Some of this white solid was dissolved in pentane from which colorless crystals suitable for X-ray analysis formed. Anal. calcd for C24H26O3Sn: C, 59.91; H, 5.45; found: C, 59.10; H, 5.42. IR (Nujol): ν/cm−1 3067 m, 3040 m, 2980 m, 1710 br vs, 1481 m, 1447 w, 1439 s, 1375 m, 1335 w, 1302 m, 1235 br s, 1150 br s, 1076 s, 1057 m, 1023 m, 997 m, 941 w, 859 w, 799 vw, 729 s, 698 s, 659 m, 540 w, 492 w, 450 w.1H NMR (500 MHz, benzene-d6): δ 0.68 (t, OCH2Me, 3H), 1.40 (d, SnOCHMe, 3H), 3.64 (q, OCH2Me, 2H), 4.64 [q, SnOCHMe, 3H 119/117Sn satellites JSnH (117Sn) 54, (119Sn) 40 Hz], 7.23 (t, m-H, 6H), 7.17 (d, p-H, 3H), 7.91 [dd, o-H, 6H JHH 4.6 and 1.9 Hz, 119/117Sn satellites JSnH (117Sn) 64, (119Sn) 48 Hz].13C{1H} NMR (126 MHz, benzene-d6)
δ: 13.81 (s, OCH2Me), 22.99 (s, SnOCHMe), 61.76 (s, OCH2Me), 68.99 (s, SnOCH2Me, 119/117Sn satellites JSnC 34 Hz), 128.72 (s, m-C, 119/117Sn satellites JSnC 60 Hz), 129.49 (s, p-C 119/117Sn satellites JSnC 13 Hz), 137.31 (s, o-C, 119/117Sn satellites JSnC 57 Hz), 142.52 (s, ipso-C), 181.13 [s, SnOCH(Me)C(O)].119Sn NMR (187 MHz, benzene-d6): δ
−143 (s).
X-Ray crystallography
The crystal of 1 was a colorless plate while 2 was a colorless chunk. Examination of the diffraction patterns on a Nonius Kappa CCD diffractometer indicated a triclinic crystal system for 1 and a monoclinic crystal system for 2. All data collection was performed at 200 K using an Oxford Cryosystems Cryostream Cooler. For 1 the data collection strategy was set up to measure a hemisphere of reciprocal space with a redundancy factor of 3.0, meaning that 90% of the reflections were measured at least 3.0 times. For 2 the data collection strategy was set up to measure a quadrant of reciprocal space with a redundancy factor of 3.7, meaning that 90% of the reflections were measured at least 3.7 times. A combination of phi and omega scans with a frame width of 1.0° was used. Data integration was done with Denzo13 whilst scaling and merging of the data was performed with Scalepack.13 Merging the data and averaging the symmetry equivalent reflections resulted in a Rint value of 0.030 for both data sets.The structures were solved by the Patterson method in SHELXS-8614 in space groups P and P21/c for 1 and 2, respectively. For 2 one of the phenyl rings is rotationally disordered about the Sn–C(19) bond. The disorder is modelled with two orientations for this ring. The C(19) and C(22) atoms are common to both orientations, while the other four carbon atoms of the ring are disordered over two sites each. Only the C(19) atom of this phenyl group is refined anisotropically; the other carbon atoms are kept isotropic. The occupancy factors for the two orientations refined to 0.529(8) and 0.471(8). Full-matrix least-squares refinements based on F2 were performed in SHELXL-93.15
For each methyl group, the hydrogen atoms were added at calculated positions using a riding model with U(H)=1.5·U(eq) (bonded C atom) for 1 and 2. The torsion angle, which defines the orientation of the methyl group about the C–C or N–C bond, was refined. The other hydrogen atoms were included in the model at calculated positions using a riding model with U(H)=1.2·U(eq) (bonded C atom). For 1 the final refinement cycle was based on 5108 intensities and 256 variables and resulted in agreement factors of R1(F)=0.031 and wR2(F2)=0.055. For 2 the final refinement cycle was based on 5704 intensities and 280 variables and resulted in agreement factors of R1(F)=0.045 and wR2(F2)=0.098. For the subset of data with I>2σ(I), the R1(F) value is 0.024 for 4503 reflections for 1 and 0.035 for 4678 reflections for 2. The final difference electron density map contains maximum and minimum peak heights of 0.77 and −0.56 e Å−3 for 1 and 0.71 and −0.91 e Å−3 for 2. Neutral atom scattering factors were used and include terms of anomalous dispersion.16 A summary of the X-ray data is given in Table 1.§
Acknowledgements
The authors wish to acknowledge the financial support from the Department of Energy, Office of Basic Sciences, Chemistry Division.References
- (a) R. Langer, Science, 2001, 293, 58 CrossRef CAS; (b) A. Lendlein and R. Langer, Science, 2002, 296, 1673 CrossRef; (c) R. Langer, Nature (London), 1998, 392, 5 CAS; (d) J. T. Santini, Jr, A. C. Richards, R. Scheidt, M. J. Cima and R. Langer, Angew. Chem., Int. Ed., 2000, 39, 2396 CrossRef; (e) R. Langer, Mol. Therapy, 2000, 1, 12 Search PubMed; (f) A. Tullo, Chem. Eng. News, 2000, 21 Search PubMed.
-
(a) T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 4072 CrossRef CAS;
(b) C. P. Radano, G. L. Baker and M. R. Smith, J. Am. Chem. Soc., 2000, 122, 1552 CrossRef CAS;
(c) M. Cheng, A. B. Attygalle, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 11584 CAS;
(d) M. H. Chisholm, J. C. Huffman and K. Phomphrai, J. Chem. Soc., Dalton Trans, 2001, 222 RSC;
(e) M. H. Chisholm, N. W. Eilerts, J. C. Huffman, S. S. Iyer, M. Pacold and K. Phomphrai, J. Am. Chem. Soc., 2000, 122, 11845 CrossRef CAS;
(f) A. P. Dove, V. C. Gibson, E. L. Marshall, A. J. P. White and D. J. Williams, Chem. Commun., 2001, 283 RSC;
(g) B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229 CrossRef CAS;
(h) M. H. Chisholm, J. Gallucci and K. Phomphrai, Inorg. Chem., 2002, 41, 2785 CrossRef CAS;
(i) T. M. Ovitt and G. W. Coates, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4686 CrossRef CAS;
(j) T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316 CrossRef CAS.
- B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans, 2001, 2215 RSC.
- Ecochem is a polylactide based packaging material developed by DuPont ConAgra.
- Leupron Depot is a product of Takeda Chemical Industries, Ltd., Japan for drug delivery purposes.
-
(a)
E. E. Schmitt and R. A. Polistina, U.S. Pat. 3463158, 1969;
(b) E. J. Frazza and E. E. Schmitt, Biomed. Mater. Symp., 1970, 1, 43 Search PubMed.
- (a) J. A. Hubbell and R. Langer, Chem. Eng. News, 1995, March 13, 42 Search PubMed; (b) R. Langer and J. P. Vacanti, Science, 1993, 260, 920 CAS.
- (a) E. F. Conner, G. W. Nyce, M. Myers, A. Mock and J. L. Hedrick, J. Am. Chem. Soc., 2002, 124, 914 CrossRef CAS; (b) G. W. Nyce, T. Glauser, E. F. Conner, A. Mock, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2003, 125, 3046 CrossRef CAS.
- C. P. Radano, G. L. Baker and M. R. Smith, J. Am. Chem. Soc., 2000, 122, 1552 CrossRef CAS.
- (a) M. H. Chisholm and E. E. Delbridge, Chem. Commun., 2001, 1308 RSC; (b) M. H. Chisholm and E. E. Delbridge, New J. Chem., 2003, 27, 1167 RSC.
- C. Camacho-Camacho, R. Contreras, H. Nöth, M. Bechmann, A. Sebald, W. Milius and B. Wrackmeyer, Magn. Reson. Chem., 2002, 40, 31 CrossRef CAS.
- E. L. King, Int. J. Chem. Kinet., 1982, 14, 1285 CAS.
- DENZO: Z. Otwinowski and W. Minor, in Macromolecular Crystallography (vol. 276 of the Methods in Enzymology series), eds. C. W. Carter, Jr. and R. M. Sweet, Academic Press, 1997, Part A, p. 307 Search PubMed.
- SHELXS-86: G. M. Sheldrick, Acta Crystallogr., Sect. A, 1990, 46, 467 Search PubMed.
- SHELXL-93: G. M. Sheldrick, Universitāt Göttingen, Germany, 1993.
- International Tables for Crystallography, Kluwer Academic Publishers., Dordrecht, 1992, vol. C Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: kinetic data of reactions C and E in Scheme 2. See http://www.rsc.org/suppdata/nj/b3/b306700a/ |
| ‡ Current address: Chemistry Department, University of North Dakota, P.O. Box 9024, Grand Forks, ND 58203-9024, USA. E-mail: edelbridge@chem.und.edu |
| § CCDC reference numbers 210941 and 210942. See http://www.rsc.org/suppdata/nj/b3/b306700a/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2004 |