(Thia)calix[4]arene–porphyrin conjugates: novel receptors for fullerene complexation with C70 over C60 selectivity
Miroslav
Dudič
a,
Pavel
Lhoták
*a,
Ivan
Stibor
a,
Hana
Petříčková
b and
Kamil
Lang
*c
aDepartment of Organic Chemistry, Institute of Chemical Technology, Technická 5, 166 28, Prague 6, Czech Republic. E-mail: lhotakp@vscht.cz; Fax: +420-224 354 288; Tel: +420-224 354 280
bDepartment of Solid State Chemistry, Institute of Chemical Technology, Technická 5, 166 28, Prague 6, Czech Republic
cInstitute of Inorganic Chemistry, Academy of Sciences of the Czech Republic, 250 68, Řež, Czech Republic. E-mail: lang@iic.cas.cz; Fax: +420 220 941 502; Tel: +420 266 172 193
First published on 31st October 2003
Abstract
Molecular tweezers, (thia)calix[4]arene–porphyrin conjugates, are constructed from the (thia)calix[4]arene unit serving as a scaffold and from two and/or four porphyrin units. These molecules form stable complexes with fullerenes in a toluene solution and exhibit selectivity towards C70. The observed fullerene–porphyrin contacts suggest cooperative behaviour of closely separated porphyrin units attracting C60 or C70. Measurements show efficient quenching of porphyrin fluorescence emission.
Introduction
Calix[4]arenes1 and more recently thiacalixarenes2 are frequently used as the building blocks or molecular scaffolds in the design of new sophisticated molecular systems. Similarly, the porphyrins3 have many possible applications in the construction of artificial molecular receptors and/or devices since they possess useful photoactive and/or electroactive properties.Fullerenes fit nicely to preorganized cavities and form stable complexes, in particular with deep-walled cavitands, calixarenes, homotrioxacalixarenes, cyclotriveratrylenes or cyclodextrins.4 It was also shown that in the solid state or even in solutions the curved π surface of C60 is attracted to the centre of a (metallo)porphyrin ring. Consequently, numerous elegantly designed porphyrin systems have been synthesized for studying porphyrin/fullerene interactions.5
As we have recently demonstrated, the combination of (thia)calix[4]arene and porphyrin units leads to novel conjugates with complexation abilities towards anions,6 cations7 or neutral8 molecules. In this study, we present receptor molecules capable of forming stable supramolecular complexes with fullerenes C60 and C70 in toluene with high selectivity for C70 (Scheme 1).
![(Thia)calix[4]arene–porphyrin receptors.](/image/article/2004/NJ/b307988k/b307988k-s1.gif) | ||
| Scheme 1 (Thia)calix[4]arene–porphyrin receptors. | ||
Results and discussion
The receptors 1–4 were synthesized and purified following reported procedures. Hydrolysis of calixarene di- or tetraacetates with NaOH yielded corresponding carboxylic acids that were condensed with the monoamino derivative of 5,10,15,20-tetraphenylporphyrin.7 Metallation of porphyrins to corresponding Zn-porphyrins 1Zn–4Zn was achieved using zinc acetate–Et3N in anhydrous CH2Cl2.8 To evaluate the role of preorganization of the calixarene cavity and cooperative behaviour of attached porphyrin units, a model compound 5, bearing only a single porphyrin unit, was prepared.Complexation was first studied by 1H NMR titrations with fullerenes C60 and C70 in toluene-d8. Upon addition of increasing amounts of fullerene to 0.2–0.5 mM solutions of the receptors, the proton resonances of the porphyrin NH moved upfield (ca. 0.4 ppm in the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) +C70 system)
(Fig. 1). Similarly, upfield shifts, albeit not so pronounced (approx. 0.15 ppm), were observed for the β-pyrrole protons of the porphyrin moieties. The chemically-induced shifts were shown only by porphyrin protons, while all protons of the calixarene skeleton remained unchanged (Fig. 2). These results indicate direct contact between the porphyrin moiety and fullerene.5 In addition, Job plots confirmed the formation of 1 ∶ 1 complexes of the receptors with fullerenes. Assuming the 1 ∶ 1 stoichiometry, the binding isotherms constructed from induced shifts of the NH and β-pyrrole resonances for the porphyrin and metalloporphyrin moieties, respectively, were analysed by nonlinear least-squares methods giving binding constants summarized in Table 1.9,10 In contrast, there was no 1H NMR evidence of a complex formation between reference mono-porphyrin conjugate 5 and fullerenes. This suggests that the (thia)calix[4]arene skeleton does not bind fullerenes and that the preorganization of two porphyrin units on the lower rim of calixarenes is the fundamental prerequisite for complexation of fullerenes. Hence, the calixarene skeleton serves as a molecular scaffold holding porphyrins in a suitable distance corresponding roughly to the size of fullerenes.
+C70 system)
(Fig. 1). Similarly, upfield shifts, albeit not so pronounced (approx. 0.15 ppm), were observed for the β-pyrrole protons of the porphyrin moieties. The chemically-induced shifts were shown only by porphyrin protons, while all protons of the calixarene skeleton remained unchanged (Fig. 2). These results indicate direct contact between the porphyrin moiety and fullerene.5 In addition, Job plots confirmed the formation of 1 ∶ 1 complexes of the receptors with fullerenes. Assuming the 1 ∶ 1 stoichiometry, the binding isotherms constructed from induced shifts of the NH and β-pyrrole resonances for the porphyrin and metalloporphyrin moieties, respectively, were analysed by nonlinear least-squares methods giving binding constants summarized in Table 1.9,10 In contrast, there was no 1H NMR evidence of a complex formation between reference mono-porphyrin conjugate 5 and fullerenes. This suggests that the (thia)calix[4]arene skeleton does not bind fullerenes and that the preorganization of two porphyrin units on the lower rim of calixarenes is the fundamental prerequisite for complexation of fullerenes. Hence, the calixarene skeleton serves as a molecular scaffold holding porphyrins in a suitable distance corresponding roughly to the size of fullerenes.
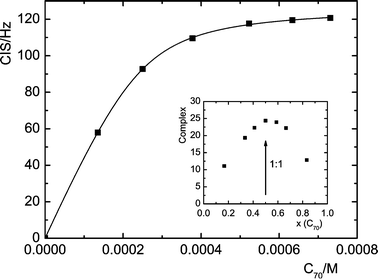 | ||
| Fig. 1 1H NMR titration of 2 (2 × 10−4 M−1) with C70 (porphyrin NH protons, 300 MHz, 298 K). The solid line is the theoretical isotherm obtained by the least-squares fit to the experimental data. Inset: Job plot for the same receptor. | ||
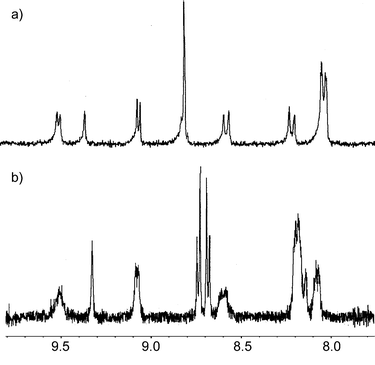 | ||
| Fig. 2 Partial 1H NMR spectra of the porphyrin signals: a) free receptor 1, b) receptor 1+5 equiv. of C70
(toluene-d8, 300 MHz, 298 K). Note splitting of the broad singlet (bs, 8H, β-pyrrole H) into two doublets. | ||
| Receptor | C60 | C70 |
|---|---|---|
|
a Experimental error 15% unless otherwise stated.
b UV–vis titration, toluene, 294 K, K/M−1: (7.6±0.9)×103 for 1-C60, (1.8±0.2)×104 for 1-C70, (3.0±0.3)×104 for 1Zn-C70.
c Insoluble in toluene.
d Line broadening.
e β-Pyrrole protons have lower CIS than NH protons, which are absent; it causes higher error of K.
|
||
| 1 | 4920 | 21100 |
| 2 | 2340 | 15600 |
| 1Zn e | 8600±1800 |
27950±7100 |
| 2Zn e | 2710±530 |
37400±9800 |
| 3 | 3510 | 3330 |
| 4 | 3420 | 6350 |
| 3Zn | c | c |
| 4Zn e | 2300±470 |
d |
| 5 | No interaction | No interaction |
| 6 | 3500 | 7920 |
| 7 | 1460 | 14500 |
In order to assess the influence of the preorganization of the calixarene unit we designed and synthesized receptors with the functionalized upper rim.6 Namely, calix[4]arene derivatives 6 and 7 were immobilised in the cone conformation with two porphyrin units being connected to the upper rim via ureido functions (Scheme 2). These calixarene–porphyrins possess similar complexation ability towards fullerenes as the receptors 1–4. The 1H NMR titration experiments revealed that compounds 6 and 7 exhibit higher complexation ability towards C70. This phenomenon is especially pronounced in the case of tetraacetate derivative 7
(cf. 1.5×103 M−1 for 7-C60 and 14.5×103 M−1 for 7-C70).
![Calix[4]arene–porphyrin receptors.](/image/article/2004/NJ/b307988k/b307988k-s2.gif) | ||
| Scheme 2 Calix[4]arene–porphyrin receptors. | ||
Further indications of complexation were obtained by UV–vis titrations. Electronic absorption spectra of 1–4 and 1Zn–4Zn have typical porphyrin features, however, the Soret bands are considerably broadened and split into at least two components when compared to a single porphyrin unit. It indicates intramolecular exciton coupling between closely separated porphyrin units due to the spatial flexibility of the amide spacer connecting them with the calixarene skeleton.6–8 After addition of C60 or C70 the original split Soret band underwent significant hypochromicity and well-defined isosbestic points appeared (Fig. 3). Evidently, the high spectral sensitivity of the receptors 1–4 and 1Zn–4Zn is due to exciton coupling since the features of the Soret band are strongly affected by interaction with fullerenes and by a porphyrin–porphyrin relative orientation. Exciton coupling does not influence the complexation because the receptors 6 and 7 with much less extent of coupling6 bind fullerenes similarly as 1–4 (Table 1, 1H NMR results). However, in this case the spectral changes are too small for quantitative interpretation. No interaction occurred between the model compounds 5 or 5,10,15,20-tetraphenylporphyrin (TPP) and fullerenes in solution since no spectral changes were observed up to 50 equiv. of fullerenes. It is noteworthy that the values of K are comparable with those obtained by 1H NMR experiments (Table 1). It demonstrates compatibility of both methods and no concentration dependent effects on the function of the receptors.
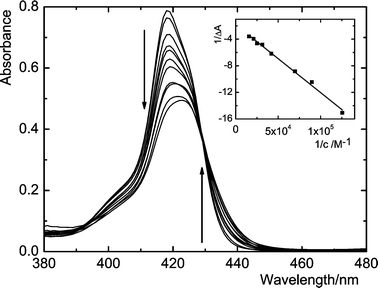 | ||
| Fig. 3 Difference UV–vis spectra showing the Soret band of 1 (1.1 μM) after addition of C70 in toluene. The isosbestic point is at 429 nm. The arrows follow changes due to increasing concentrations of C70, varied from 0 to 64 μM. Inset: Benesi–Hildebrand plot to the experimental data. | ||
The complexation was also evidenced by fluorescence spectroscopy. While the lifetime of 1Zn (1.92 ns) did not show any changes upon addition of fullerenes, steady-state fluorescence was strongly quenched (Fig. 4). Evidently, quenching is a consequence of photoinduced electron transfer between 1S of the porphyrin moiety and C70.11 The fluorescence decay–time profiles indicate that the lifetime of porphyrin in the complexes is less than 100 ps, i.e. below the time resolution of our instrument.
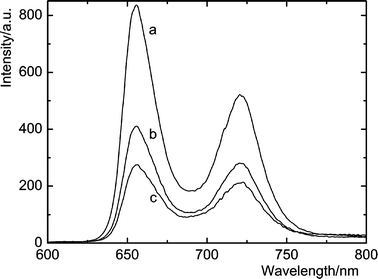 | ||
| Fig. 4 Steady-state fluorescence spectra of 1.5 μM 1 (a) in the presence of 35.8 μM (b) and 71.6 μM C70 (c) in toluene. | ||
Comparison of 1, 2, 6, 7 and model compounds (5, TPP) clearly indicates that the cooperative effect of two porphyrin units is crucial for the fullerene complexation. The UV–vis and fluorescence results reveal that the complex enables porphyrins and fullerenes to undergo electronic coupling. The preorganization of the lower or upper rim of (thia)calixarene with two cofacially oriented porphyrin moieties creates a cavity where fullerene can be inserted to form the 1 ∶ 1 complex. A substantial increase of selectivity for C70 is observed for the tetraacetate (7) over tetrapropoxy (6) calixarene although no effect was anticipated due to their similar size. The respective substituents were also reported to affect hydrogen bonding between anions and ureido functions at the upper rims of 6 and 7.6 Evidently this subtle change to the calixarene structure effectively influences the binding affinity at the opposite upper rim. It appears to be an important finding because it renders an efficient way to modulate the binding selectivity by simple functionalization of the lower rim. Introduction of four porphyrins on the lower rim of the receptors (3, 4, 4Zn) does not improve the fullerene complexation (cf. 4.9×103 M−1 for 1–C60 and 3.5×103 M−1 for 3–C60) and leads to the loss of the fullerene recognition (cf.KC70/KC60 is 4.3 and ∼1 for 1 and 3, respectively). We suppose that fullerenes are not well complemented by the four-armed calixarenes and that the stacking of the porphyrin units within the molecule constrains the four-point binding motif.
The 1H NMR study of the lower-rim substituted calixarene 1 and thiacalixarene 2 did not reveal differences in the conformational behaviour. However, a high downfield shift of the NH amidic signals (11.11 ppm for 1, 11.13 ppm for 2; chemical shifts were concentration independent) might indicate that the preorganization of 1–4 is strengthened via intramolecular hydrogen bonding. Our attempts to grow suitable monocrystals for X-ray studies of structural motifs have failed. Hence, we synthesized model compounds, 4-methylphenyl diamides 8–11 (Scheme 3), with the similar aromatic amide structural fragments. We succeeded in growing suitable crystals of 8 and 11 using slow evaporation of an ethyl acetate/ethanol solution.
 | ||
| Scheme 3 Model compounds for crystallographic study. | ||
The classical calix[4]arene 8 adopts the cone conformation where both amidic hydrogens are engaged in hydrogen bonding with neighbour oxygen atoms of the –O–CH2– and OH groups. The corresponding NH⋯O distances are in the range of 2.06–2.20 Å (Fig. 5a). The resulting hydrogen bonding array stabilizes the calixarene core with approx. C2 symmetry and contributes to the preorganization of the lower rim with two coplanar aromatic units separated by 3.54 Å (Fig. 5b).
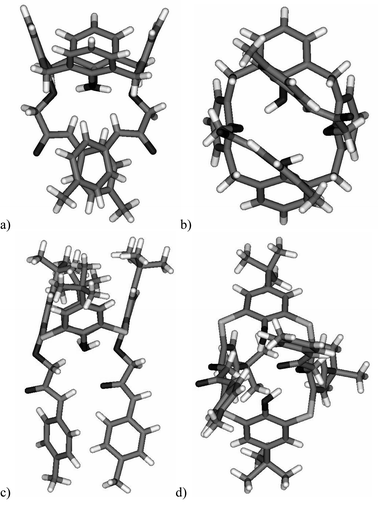 | ||
| Fig. 5 Solid-state structures of derivatives 8 (a,b) and 11 (c,d). | ||
The hydrogen bonding arrangement within thiacalixarene 11 is completely different. The NH group of an amidic unit is connected to the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group (NH⋯O distance=2.11 Å) of the diametrical amide arm (Fig. 5c). This intramolecular bonding leads to the overall unsymmetrical structure with one amide NH function facing towards the CO group of a neighbour molecule. The distance of this intermolecular hydrogen bonding (NH⋯O) is of 1.92 Å. The unsymmetrical organisation of the hydrogen bonds results in structural changes to the lower rim. Consequently, the mutual position of the two 4-methylphenyl units is no longer coplanar as within 8, but rather almost perpendicular (Fig. 5d).
O group (NH⋯O distance=2.11 Å) of the diametrical amide arm (Fig. 5c). This intramolecular bonding leads to the overall unsymmetrical structure with one amide NH function facing towards the CO group of a neighbour molecule. The distance of this intermolecular hydrogen bonding (NH⋯O) is of 1.92 Å. The unsymmetrical organisation of the hydrogen bonds results in structural changes to the lower rim. Consequently, the mutual position of the two 4-methylphenyl units is no longer coplanar as within 8, but rather almost perpendicular (Fig. 5d).
Although we cannot confirm the same hydrogen bonding patterns in the solution, these results are indicative of the specific behaviour of both molecular systems. The solid-state conformational preferences could be used as fundamentals for the explanation of calixarene vs. thiacalixarene complexation ability towards fullerenes.
Metallation of 1 and 2 (1Zn, 2Zn) does not reduce the binding constants opposed to the Boyd's experimental observation that free-base porphyrins bind C60 more strongly.5f It is clear that a number of effects can influence binding constants and the ordering of free-base and metalloporphyrins can be ascribed to subtle interplay of charge transfer, electrostatics and solvation energy effects. The solvation effects can be very important as documented by our observation that the receptors do not bind fullerenes in more polar 1,2-dichlorobenzene as follows from 1H NMR and UV–vis experiments. This can be ascribed to the fact that attractive electrostatic interactions contribute approximately 50–60% to the total attractive interactions.12
The most interesting feature is the preference of C70 over C60.13,14 For example, the receptor 2 with K of 1.6×104 M−1 and 2.3×103 M−1 for C70 and C60, respectively, gives the C70/C60 selectivity of about 7. Despite relatively flexible nature of the porphyrin tweezers, the presented receptors can efficiently differentiate between C70 and C60. The higher affinity towards C70 was suggested to be a result of the ovoid shape allowing maximization of C70–porphyrins interaction.13a
In conclusion, we have introduced novel fullerene receptors, molecular tweezers, constructed from (thia)calix[4]arene and porphyrin moieties. The complexation occurs in a toluene solution and can be quantitatively investigated using common spectroscopic methods (UV–vis, NMR, fluorescence). The selectivity towards C70 opens the way towards self-assembling systems and new separation and purification methods for fullerenes.
Experimental
Compounds
The syntheses of compounds 1–4, 6, 7 and the corresponding Zn2+ salts 1Zn–4Zn were carried out according to published procedures.6–8 The melting point is uncorrected and was determined using a Boetius Block apparatus. The 1H NMR spectra were recorded on a Varian Gemini 300 using tetramethyl silane as an internal standard.Procedure for the preparation of derivative 5
The mixture of starting monocarboxy-triethylester derivative of calix[4]arene (prepared by monohydrolysis of the corresponding tetraester15) (100 mg; 0.10 mmol) and dicyclohexylcarbodiimide (65 mg; 0.32 mmol) in CH2Cl2 (10 ml) was stirred for 10 minutes and then monoamino derivative of 5,10,15,20-tetraphenylporphyrin (63 mg; 0.10 mmol) was added. The reaction mixture was then stirred overnight at room temperature. The solvent was removed under reduced pressure, the residue was dissolved in CH2Cl2 (30 ml) and washed with water (30 ml). The organic layer was dried over MgSO4. The crude product was purified by column chromatography on silica gel using CHCl3–petroleum ether mixture as an eluent to yield the product 5 (81 mg, 51%). Mp 185–188°C. 1H NMR (CDCl3, 300 MHz)
δ: −2.77 (s, 2H, NH por.), 1.04 (s, 9H, But), 1.07 (s, 9H, But), 1.18 (s, 18H, But), 1.13 (m, 9H, –OCH2CH3), 3.29 (d, J=12.80 Hz, 2H, eq. ArCH2Ar), 3.76 (d, J=13.60 Hz, 2H, eq. ArCH2Ar), 4.05–4.24 (m, 6H, –OCH2CH3), 4.72–4.84 (m, 8H, ax. ArCH2Ar and –COCH2–), 4.97 (d, J=12.80 Hz, 2H, ax. ArCH2Ar), 5.17 (d, J=16.50 Hz, 2H, –COCH2–), 6.79 (d, J=6.20 Hz, 4H, H-arom.), 6.95 (dd, J=11.00 Hz, J=2.20 Hz, 4H, H-arom.), 7.77 (m, 9H, H-arom.), 8.22 (m, 10H, H-arom.), 8.86 (m, 6H, H-arom.), 8.93 (d, J=5.10 Hz, 2H, H-arom.), 10.02 (s, 1H, –CONH–). IR (CHCl3)
νmax
(cm−1): 1754, 1683 (CO), 3324 (NH). FAB MS m/z
(rel. int.) 1576 [M+1]+
(100).
Preparation of model amides 8–11
The mixture of starting dicarboxymethyl derivative of (thia)calix[4]arene (prepared by hydrolysis of the corresponding diesters7) (0.26 mmol) and dicyclohexylcarbodiimide (1.04 mmol) in CH2Cl2 (10 ml) was stirred for 15 minutes at room temperature and then 4-methylaniline (0.57 mmol) was added. The reaction mixture was stirred overnight, and the solvent was removed under reduced pressure. The crude product was purified by column chromatography on silica gel using a CHCl3–petroleum ether mixture as an eluent to yield the products 8–11.
8 Yield: 56%, white crystals, mp: 304–306°C (ethyl acetate). 1H NMR (300 MHz, CDCl3)
δ: 2.30 (s, 6H, CH3Ar), 3.57 (d, J=13.50 Hz, 4H, eq. ArCH2Ar), 4.23 (d, J=13.20 Hz, 4H, ax. ArCH2Ar), 4.62 (s, 4H, OCH2CO), 6.78 (t, J=7.70 Hz, 2H, H-arom.), 6.90 (t, J=6.90 Hz, 2H, H-arom.), 7.02 (t, J=8.80 Hz, 8H, H-arom.), 7.15 (d, J=7.40 Hz, 4H, H-arom.) 7.25 (d, J=8.30 Hz, 4H, H-arom.), 8.30 (s, 2H, ArOH), 10.13 (s, 2H, –CONH). FAB MS m/z
(rel. int.) 719 [M+1]+
(100).
9 Yield: 76%, white crystals, mp: 160–162°C (ethyl acetate–ethanol 4 ∶ 1). 1H NMR (300 MHz, CDCl3)
δ: 1.08 (s, 18H, But), 1.28 (s, 18H, But), 2.29 (s, 6H, CH3Ar), 3.51 (d, J=13.70 Hz, 4H, eq. ArCH2Ar), 4.22 (d, J=13.20 Hz, 4H, ax. ArCH2Ar), 4.60 (s, 4H, OCH2CO), 6.98 (d, J=6.80 Hz, 4H, H-arom.), 6.99 (s, 4H, H-arom.), 7.12 (s, 4H, H-arom.), 7.27 (d, J=6.80 Hz, 4H, H-arom.), 7.71 (s, 4H, H-arom.), 8.09 (s, 2H, ArOH), 10.15 (s, 2H, –CONH). FAB MS m/z
(rel. int.) 943 [M+1]+
(100).
10. Yield: 65%, colourless crystals, mp: decomp. 280–282°C (CHCl3–MeOH). 1H NMR (300 MHz, CDCl3)
δ: 2.31 (s, 6H, CH3Ar), 4.75 (s, 4H, OCH2CO), 6.85 (t, J=8.00 Hz, 2H, H-arom.), 6.94 (t, J=7.70 Hz, 2H, H-arom.), 7.05 (d, J=8.25 Hz, 4H, H-arom.), 7.33 (d, J=8.00 Hz, 4H, H-arom.) 7.46 (d, J=7.70 Hz, 4H, H-arom.), 7.72 (d, J=7.70 Hz, 4H, H-arom.), 8.49 (s, 2H, ArOH), 10.05 (s, 2H, –CONH). FAB MS m/z
(rel. int.) 791 [M]+
(100).
11 Yield: 72%, colourless crystals, mp: 251–252°C (CHCl3–ethanol 5 ∶ 1). 1H NMR (300 MHz, CDCl3)
δ: 1.09 (s, 18H, But), 1.29 (s, 18H, But), 2.30 (s, 6H, CH3Ar), 4.70 (s, 4H, OCH2CO), 7.05 (d, J=8.30 Hz, 4H, H-arom.), 7.36 (d, J=8.50, 4H, H-arom.), 7.50 (s, 4H, H-arom.), 7.71 (s, 4H, H-arom.), 8.54 (s, 2H, ArOH), 10.18 (s, 2H, –CONH). FAB MS m/z
(rel. int.) 1015 [M+1]+
(100).
Crystallographic study
X-Ray data for 8: C46H42O6N2: Mr=718.85, monoclinic system, space group P 21/n, a=15.3648(13)
Å, b=14.4941(14)
Å, c=16.7893(11)
Å, β=91.015(6)°, V=3738.4(5)
Å3, Z=4, Dc=1.28 g·cm−3, μ(CuKα)=0.678 cm−1, crystal size 0.1×0.3×0.4 mm. Data were measured at 293 K on an Enraf-Nonius CAD4 diffractometer with graphite monochromated CuKα radiation (λ=1.5418Å). The structure was solved by direct methods,16 oxygen and nitrogen atoms were anisotropically and carbons isotropically refined by full matrix least-squares on F values17 to final R=0.0910, Rw=0.0810 and S=1.178 with 257 parameters using 2093 independent reflections (θmax=67.98°). Hydrogen atoms linked to carbon atoms were located from expected geometry and were not refined. Hydrogens linked to oxygen and nitrogen atoms were found from Fourier difference electron density map and their position was not refined. CCDC reference number 220090. See http://www.rsc.org/suppdata/nj/b3/b307988k/ for crystallographic data in .cif or other electronic format.
X-Ray data for 11: C58H66N2O6S4·C2H6O: Mr=1057.47, monoclinic system, space group P 21/c, a=18.037(3)
Å, b=17.973(2)Å, c=19.697(2)Å, β=112.25(1)°, V=5910.0(13)Å3, Z=4, Dc=1.19 g·cm−3, μ(CuKα)=1.881 cm−1, crystal size 0.05×0.4×0.8 mm. Data were measured at 293 K on an Enraf-Nonius CAD4 diffractometer with graphite monochromated CuKα radiation (λ=1.5418Å). The structure was solved by direct methods,16 sulfur and nitrogen atoms were anisotropically and oxygen and carbon atoms refined by full matrix least-squares on F values17 to final R=0.113, Rw=0.101 and S=1.170 with 280 parameters using 2500 independent reflections (θmax=69.95°). Hydrogen atoms were located from expected geometry and were not refined. Hydrogen atoms linked to the oxygen and nitrogen atoms were not found. Three of four t-Bu groups were disorder so that the model of disorder was used. CCDC reference number 220091. See http://www.rsc.org/suppdata/nj/b3/b307988k/ for crystallographic data in .cif or other electronic format.
Spectral measurements
Absorption spectra were measured on a Perkin-Elmer Lambda 35 spectrometer. Titrations were carried out by stepwise addition of fullerenes dissolved in toluene to a toluene solution of 1–2 μM receptor and recorded with the same fullerene concentration in the reference position. Spectral data were cast into the double-reciprocal form and analysed using the Benesi–Hildebrand equation.9 All experiments were performed in toluene at 21°C.
The fluorescence spectra were recorded on a Perkin-Elmer LS 50B luminescence spectrophotometer. All emission spectra were corrected for the characteristics of the detection monochromator and photomultiplier. The absorbances of 1 and TPP were adjusted to the same value at the excitation wavelength of 515 nm. Because TPP does not interact with the receptors the inner filter effect due to added fullerene can be eliminated by comparison of intensity of the receptor with that of TPP.
The binding constants were assessed from the 1H NMR titration experiments using initial concentrations of the receptors ranging from 0.2 to 0.5 mM. The concentration of fullerene C60 or C70 was gradually increasing to cover the range of saturation up to 90%. The induced chemical shifts of NH signals were recorded for 1–7. Due to the absence of the NH signals in Zn-derivatives 1Zn–4Zn, the shifts of the β-pyrrole protons were plotted against the concentration of fullerene to construct titration curves. Titration data were analysed using the original non-linear regression curve-fitting computer program OPIUM.10
Acknowledgements
The authors wish to thank the Grant Agency of the Czech Republic for financial support of this work (GA 203/03/0926).References
- For books on calixarenes see: (a) Calixarenes 2001, eds. Z. Asfari, V. Böhmer, J. Harrowfield and J. Vicens, Kluwer Academic Publishers, Dordrecht, 2001 Search PubMed; (b) Calixarenes in Action, eds. L. Mandolini and R. Ungaro, Imperial College Press, London, 2000 Search PubMed; (c) C. D. Gutsche, Calixarenes Revisited: Monographs in Supramolecular Chemistry, ed. J. F. Stoddart, The Royal Society of Chemistry, Cambridge, 1998, Vol. 6 Search PubMed; (d) Calixarenes 50th Anniversary: Commemorative Issue, eds. J. Vicens, Z. Asfari and J. M. Harrowfield, Kluwer Academic Publishers, Dordrecht, 1994 Search PubMed; (e) Calixarenes: A Versatile Class of Macrocyclic Compounds, eds. J. Vicens and V. Böhmer, Kluwer Academic Publishers, Dordrecht, 1991 Search PubMed.
- H. Kumagai, M. Hasegawa, S. Miyanari, Y. Sugawa, Y. Sato, T. Hori, S. Ueda, H. Kamiyama and S. Miyano, Tetrahedron Lett., 1997, 38, 3971 CrossRef CAS.
- J. L. Sessler, B. Wang, S. L. Springs and C. T. Brown, in Comprehensive Supramolecular Chemistry, ed. Y. Murakami, Elsevier Science Ltd., Oxford, 1996, Vol. 4, p. 311 Search PubMed.
- (a) F. C. Tucci, D. M. Rudkevich and J. Rebek, Jr., J. Org. Chem., 1999, 64, 4555 CrossRef CAS; (b) J. L. Atwood, G. A. Koutsantonis and C. L. Raston, Nature, 1994, 368, 229 CrossRef CAS; (c) S. Suzuki, N. Nakashima and S. Shinkai, Chem. Lett., 1994, 699 CAS; (d) J. L. Atwood, L. J. Barbour, C. L. Raston and I. B. N. Sudria, Angew. Chem., Int. Ed., 1998, 37, 981 CrossRef CAS; (e) M. J. Hardie and C. L. Raston, Chem. Commun., 1999, 1153 RSC; (f) M. Yanase, T. Haino and Y. Fukazawa, Tetrahedron Lett., 1999, 40, 2781 CrossRef CAS; (g) J. Wang, S. G. Bodige, W. H. Watson and C. D. Gutsche, J. Org. Chem., 2000, 65, 8260 CrossRef CAS; (h) A. M. Bond, W. Miao, C. L. Raston and C. A. Sandoval, J. Phys. Chem. B, 2000, 104, 8129 CrossRef CAS; (i) S. Mizyed, M. Ashram, D. O. Miller and P. E. Georghiou, J. Chem. Soc., Perkin Trans. 2, 2001, 1916 RSC; (j) C. N. Murthy and K. E. Geckeler, Chem. Commun., 2001, 1194 RSC; (k) A. Buvári-Barcza, J. Rohonczy, N. Rozlosnik, T. Gilányi, B. Szabo, G. Lovas, T. Braun, J. Samu and L. Barcza, J. Chem. Soc., Perkin Trans. 2, 2001, 191 RSC; (l) T. Haino, H. Araki, Y. Yamanaka and Y. Fukazawa, Tetrahedron Lett., 2001, 42, 3203 CrossRef CAS; (m) M. Yanase, M. Matsuoka, Y. Tatsumi, M. Suzuki, H. Iwamoto, T. Haino and Y. Fukazawa, Tetrahedron Lett., 2001, 42, 493 CrossRef; (n) J. L. Atwood, L. J. Barbour, M. W. Heaven and C. L. Raston, Chem. Commun., 2003, 2270 RSC; (o) J. L. Atwood, L. J. Barbour, M. W. Heaven and C. L. Raston, Angew. Chem., Int. Ed., 2003, 42, 3254 CrossRef.
-
(a) K. Tashiro, T. Aida, J. Y. Zheng, K. Kinbara, K. Saigo, S. Sakamoto and K. Yamaguchi, J. Am. Chem. Soc., 1999, 121, 9477 CrossRef CAS;
(b) P. D. W. Boyd, M. C. Hodgson, C. E. F. Rickard, A. G. Oliver, L. Chaker, P. J. Brothers, R. D. Bolskar, F. S. Tham and C. A. Reed, J. Am. Chem. Soc., 1999, 121, 10487;
(c) D. Sun, F. S. Tham, C. A. Reed, L. Chaker, M. Burgess and P. D. W. Boyd, J. Am. Chem. Soc., 2000, 122, 10704 CAS;
(d) T. Nishioka, K. Tashiro, T. Aida, J. Y. Zheng, K. Kinbara, K. Saigo, S. Sakamoto and K. Yamaguchi, Macromolecules, 2000, 33, 9182 CrossRef CAS;
(e) D. M. Guldi, T. Da Ros, P. Braiuca, M. Prato and E. Alessio, J. Mater. Chem., 2001, 12, 2001 Search PubMed;
(f) D. Sun, F. S. Tham, C. A. Reed, L. Chaker and P. D. W. Boyd, J. Am. Chem. Soc., 2002, 124, 6604 CrossRef CAS;
(g) M. Ayabe, A. Ikeda, S. Shinkai, S. Sakamoto and K. Yamaguchi, Chem. Commun., 2002, 1032 RSC;
(h) D. V. Konarev, A. Y. Kovalevsky, X. Li, I. S. Neretin, A. L. Litvinov, N. V. Drichko, Y. L. Slovokhotov, P. Coppens and R. N. Lyubovskaya, Inorg. Chem., 2002, 41, 3638 CrossRef CAS;
(i) Y. Sun, T. Drovetskaya, R. D. Bolskar, R. Bau, P. D. W. Boyd and C. A. Reed, J. Org. Chem., 1997, 62, 3642 CrossRef CAS.
- M. Dudic, P. Lhoták, I. Stibor, K. Lang and P. Proskova, Org. Lett., 2003, 5, 149 CrossRef CAS.
- M. Dudic, P. Lhoták, I. Stibor, H. Dvoráková and K. Lang, Tetrahedron, 2002, 58, 5475 CrossRef CAS.
- M. Dudic, P. Lhoták, H. Petricková, I. Stibor, K. Lang and J. Sykora, Tetrahedron, 2003, 59, 2409 CrossRef CAS.
- K. A. Connors, Binding constants, The measurement of molecular complex stability, John Wiley & Sons, New York, 1987 Search PubMed.
- The binding constants were calculated using the computer program OPIUM (M. Kyvala). This software is freely available at http://www.natur.cuni.cz/∼kyvala/opium.html.
- D. M. Guldi, C. Luo, M. Prato, E. Dietel and A. Hirsch, Chem. Commun., 2000, 373 RSC.
- Y. B. Wang and Z. Lin, J. Am. Chem. Soc., 2003, 125, 6072 CrossRef CAS.
- (a) J. Y. Zheng, K. Tashiro, Y. Hirabayashi, K. Kinbara, K. Saigo, T. Aida, S. Sakamoto and K. Yamaguchi, Angew. Chem., Int. Ed., 2001, 40, 1858; (b) T. Haino, M. Yanase and Y. Fukazawa, Angew. Chem. Int. Ed., 1998, 37, 997 CrossRef CAS; (c) T. Haino, H. Araki, Y. Fujiwara, Y. Tanimoto and Y. Fukazawa, Chem. Commun., 2002, 2148 RSC; (d) H. Matsubara, S. Oguri, K. Asano and K. Yamamoto, Chem. Lett., 1999, 431 CrossRef CAS.
- N. Komatsu, Org. Biomol. Chem., 2003, 1, 204 RSC.
- M. A. McKervey, E. M. Seward, G. Ferguson, B. Ruhl and S. J. Harris, J. Chem. Soc., Chem. Commun., 1985, 388 RSC.
- A. Altomare, G. Cascarano, G. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, SIR92 - a program for automatic solution of crystal structures by direct methods. J. Appl. Crystallogr. 1994, 27, 435 Search PubMed.
- D. J. Watkin, C. K. Prout, J. R. Carruthers, P. W. Betteridge and R. I. Cooper, 2001, CRYSTALS, Issue 11. Chemical Crystallography Laboratory, Oxford, UK.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2004 |