4-Ethynylpyridine as bridging moiety in mixed Ru/Re complexes
Fritz E.
Kühn
*a,
Jing-Lin
Zuo
a,
Fabrizia
Fabrizi de Biani
b,
Ana M.
Santos
ac,
Yanmei
Zhang
a,
Jin
Zhao
a,
Angela
Sandulache
a and
Eberhardt
Herdtweck
a
aAnorganisch-Chemisches Institut der Technischen Universität München, Lichtenbergstraße 4, D-85747, Garching bei München, Germany. E-mail: fritz.kuehn@ch.tum.de; Tel: 0049-89-28913108
bDipartimento di Chimica dell'Universita di Siena, Via Aldo Moro, 53100, Siena, Italy
cInstituto de Tecnologia Química e Biológica da Universidade Nova de Lisboa, Quinta do Marquês, EAN, Apt 127, 2781-901, Oeiras, Portugal
First published on 29th October 2003
Abstract
Two ruthenium(II) 4-ethynylpyridine-hydride complexes bearing one Lewis-basic nitrogen atom as coordination site, namely trans-Ru(dppe)2H(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Cpy-4)
(1) and trans-Ru(dppm)2H(CCpy-4)
(2)
(dppe
Cpy-4)
(1) and trans-Ru(dppm)2H(CCpy-4)
(2)
(dppe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =1,2-bis(diphenylphosphino) ethane, dppm=1,2-bis(diphenylphosphino) methane) and Ru(dmpe)2(CCpy-4)2
(3)
(dmpe=1,2-bis(dimethylphosphino) ethane) with two nitrogen donor atoms were applied to synthesize the heterobimetallic mixed Ru/Re complexes 4–7. The X-ray crystal structure of the binuclear complex [Re(CO)3(t-bu2bipy)Ru(dppe)2(CCpy-4)H]
[OS(
=1,2-bis(diphenylphosphino) ethane, dppm=1,2-bis(diphenylphosphino) methane) and Ru(dmpe)2(CCpy-4)2
(3)
(dmpe=1,2-bis(dimethylphosphino) ethane) with two nitrogen donor atoms were applied to synthesize the heterobimetallic mixed Ru/Re complexes 4–7. The X-ray crystal structure of the binuclear complex [Re(CO)3(t-bu2bipy)Ru(dppe)2(CCpy-4)H]
[OS(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)2CF3]
(t-bu2bipy=4, 4′-di (t-butyl)-2,2′-bipyridine, (7)) has been determined.
O)2CF3]
(t-bu2bipy=4, 4′-di (t-butyl)-2,2′-bipyridine, (7)) has been determined.
Besides the usual characterization (IR, NMR, UV/Vis, EA) of the compounds 1–7, thermogravimmetry (TG) and cyclovoltammetry (CV) of selected complexes were measured in order to study the interaction between the organometallic building blocks. The existence of long-range Ru⋯Re interactions has been observed.
Introduction
The development of molecular rods and wires as well as model complexes for their subunits has attracted considerable attention due to the potential applications of such rods in the fields of molecular electronics and devices and due to the ubiquitous role of oligomers and polymers of this type in electronics and optoelectronics.1,2 It is noteworthy that polymers and oligomers of organometallic complexes may also display novel conductive and magnetic properties.3 Due to the large variety of structures and electronic states of organometallic fragments, we are interested in the incorporation of transition metals into model complexes for oligomers and polymers. Metal σ-acetylide derivatives have found applications in material science due to their linear arrays and delocalized π-systems.4,5 They are also implicated in several important organometallic processes, such as cycloaddition reactions and catalytic dimerization of terminal acetylenes.6 After using complexes of this type for linking compounds with metal–metal bonds along the propagation axis,7 we are now exploring the possibility of employing the ruthenium(II) hydride σ-pyridylacetylide complexes, trans-RuL2H(CCpy-4)
(L=1,2-bis(diphenylphosphino) ethane, dppe, 1; 1,2-bis(diphenylphosphino) methane, dppm, 2), and the bis(σ-pyridylacetylide) complex, trans- Ru(dmpe)2(CCpy-4)2
(3)
(dmpe=1,2-bis(dimethylphosphino)ethane) as models for the synthesis of bimetallic Ru/Re compounds with the Re centres in the formal oxidation state I to examine the Ru–Re interaction more closely. To the best of our knowledge very little work on related complexes has been performed and reported to date.4e,7a As Re starting material [Re(N–N)(CO)3(MeCN)]OTf (N–N=bipy, t-bu2bipy, OTf=F3C-SO3−) has been selected. This particular compound and some derivatives have gained significant attention during the last decade. Molecules of the type fac-[L]Re(CO)3(L′)n+
(L=bipyridine, 1,10-phenathroline and its substituted derivatives; L′=Cl, pyridine and derivatives, PR3, NCCH3, etc.) are discussed as sensitizers in solar energy storage and as models for intramolecular electron transfer reactions.8 The latter detailed examinations are the reason for the application of [Re(N–N)(CO)3(MeCN)]OTf in this work.
Results and discussion
Preparation and characterization of ruthenium alkynyl-hydride and bis(σ-pyridylacetylide) complexes
Acetylide complexes of transition metals have been prepared by the replacement of metal-coordinated ligands by acetylides or acetylide derivatives,9 by deprotonation of vinylidene species,10 by insertion of a co-ordinatively unsaturated metal center into the C–H bond of a terminal acetylene,11 by protonation of hydroxo complexes with terminal alkynes,12 or by the reaction of alkynylstannanes with transition metal centers.13 More recently, ruthenium bis(σ-acetylide) complexes have also been synthesized by the reaction of [RuL2Cl2] or [RuL2H2] with terminal acetylenes in methanol solution.14 In view of the fact that the pyridine ligand is capable of ligation to a metal fragment and leads to the formation of multinuclear or even polymeric metal systems, 4-ethynylpyridine has been used as starting material to synthesize ruthenium mono or bis(σ-pyridylacetylide) complexes.
Trans-Ru(dppe)2H(CCpy-4)
(1) and trans-Ru(dppm)2H(CCpy-4)
(2) are yellow solids that are readily prepared from the reaction of cis-RuL2Cl2
(L=dppe, dppm) with sodium and 4-ethynylpyridine in methanol (eqn. (1)). This is a convenient and effective route to synthesize hydride acetylide complexes, which are assumed to be important intermediates in the process of alkyne–vinylidene tautomerization promoted by transition metal complexes.15 The resulting pale yellow solids are air stable, diamagnetic and soluble in all common non-polar organic solvents.
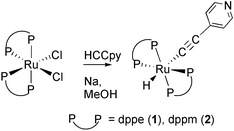 | (1) |
The 31P{1H} NMR spectra of the complexes 1 and 2 exhibit one singlet (δ=67.7 ppm for 1 and δ=2.3 ppm for 2), which suggests an equatorial arrangement of phosphine ligands, with the H− and alkynyl group being trans-disposed in both cases. The 13C-NMR spectra of compounds 1, (2), however, show two sets of signals separated by ca. 1.5, (3), 0.2, (1), 0.1, (0.3), and 0.05, (0.1) ppm for the phenyl α, ß, γ, and δ-carbon atoms, respectively, of the dppe, (dppm) groups. The bridging carbon atoms of the diphosphine ligands (dppm/dppe) are split into multiplets. This signal separation must originate from the phenyl groups on the hydride side of the molecule and of the 4-ethynylpyridyl side of the molecule, which are not equivalent. The reason for the different magnitude of the separation of the phenyl groups may be due to the proximity of the metal-bond hydrogen to the respective carbon atoms. This interpretation is supported by the 1H-NMR spectra of compounds 1 and 2. Both show two signal sets for the hydrogens riding on the bridging carbon atoms between the two phosphorus atoms in both dppe and dppm. In the case of compound 1 the signals are observed at 2.43 and 1.96 ppm (in each case the signal size is equivalent to four hydrogen atoms), in the case of compound 2 the signals are observed at 4.78 and 4.53 ppm (the signal size is equivalent to two hydrogen atoms each). The two different signal sets stem from the hydrogens above and below the P4Ru plane, which are on the side of the hydride and of the ethynylpyridine, respectively. The dppm and dppe phenyl protons are split in complicated multiplets, spanning over a much wider region than the respective 1H-NMR signals of the free ligands. The ethynylpyridine carbons are – of course - not split in two sets. The 1H NMR spectra furthermore show the presence of the hydride ligands, displaying the quintets at high field (δ(1H)=−9.71 ppm and JPH=22.0 Hz in 1; δ(1H)=−6.30 ppm, JPH=19.9 Hz in 2).
The Ru–H stretching frequencies in the IR spectra are located at 1823 cm−1 for 1 and 1772 cm−1 for 2. The asymmetric vibrations νasym, CC for 1 and 2 are observed at 2055 and 2066 cm−1, respectively. The NC-pyridine vibration occurs at 1584 cm−1 in both cases. In HCC–py νCC is located at 2098 cm−1, the NC-pyridine band is observed at 1592 cm−1. The FAB-MS spectrum of compound 1 shows the [M]+ peak at 1005 amu with a relative intensity of 20%, the [M–(CC–py)]+ peak (903 amu) is the most intense peak of the spectrum (100%). Under FAB conditions the Ru–CC–py bond therefore is the easiest to break.
The isomerisation processes of hydrido-alkynyl to vinylidene complexes (eqn. (2)) in complexes 1 and 2 are probably inhibited by the basicity of the pyridine moiety or the steric effect of dppm and dppe ligands, so that both complexes are stable in solid state at room temperature. In solution phase, the main products are the hydrido-alkynyl isomers. However, tiny amounts of the corresponding vinylidene complexes can be observed forming over time. This fact was evidenced by time-dependent UV, 1H, 31P, and 13C NMR spectroscopy.
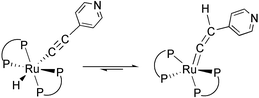 | (2) |
We were unable to prepare ruthenium bis(σ-pyridylacetylide) complexes with dppe and dppm ligands even using a five-fold excess of 4-ethynylpyridine. This is very likely due to the steric effects of the dppm and dppe ligands as they prevent the further reaction of the second hydride with 4-ethynylpyridine. For the less bulky ligand, dmpe, the bis(acetylide) complex 3 has been successfully prepared from [Ru(dmpe)2H2] by reaction with 4-ethynylpyridine in methanol solution (eqn. (3)). It displays a single sharp singlet resonance at δ(31P{1H})=38.9 ppm (pyridine–acetylide ligands in a trans-configuration around the ruthenium center). The resonance of the metal-bound carbon in the acetylide ligand appears at δ(13C)=144.6 ppm, the pyridine-bound ethynyl carbon atom appears at δ(13C)=109.5 ppm. The 13C-NMR signals of the dmpe carbon atoms are not split into two sets of signals, due to the symmetry of the molecule 3 to the P4Ru mirror plane. Accordingly, there is only one proton signal in the 1H-NMR spectrum for the ethylene bridge between the dmpe phosphorus atoms. The stretching frequency of CC bond is located at 2052 cm−1, which is similar to other bis(acetylide) complexes of ruthenium.13,14
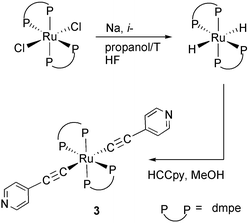 | (3) |
Trinuclear units with ruthenium acetylide and rhenium carbonyl complexes as building blocks
Treatment of 3 with two equivalents of [Re(N–N)(CO)3(MeCN)]OTf under reflux in THF afforded the trimetallic complexes [{Re(N–N)(CO)3}2{μ-(4-pyCC)2Ru(dmpe)2}](OTf)2
(N–N=2,2′-bipyridine, bipy, 4; 4, 4′-di (t-butyl)-2, 2′-bipyridine, t-bu2bipy, 5) according to eqn. (4). They are air and thermally stable yellow solids. Complex 5 is soluble in most common organic solvents, but complex 4 is only moderately soluble in CH2Cl2 and soluble in CH3CN and acetone.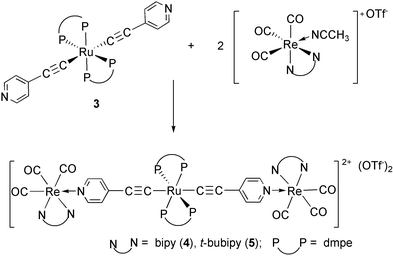 | (4) |
The spectroscopic properties of the new complexes are consistent with the proposed formulae. In the 31P{1H} NMR spectra, a single sharp singlet resonance appears at δ=37.7 ppm in complex 4 and δ=37.6 ppm in complex 5, showing a slight up-field shift compared with the chemical shift of the free ruthenium precursor 3
(38.9 ppm). The 13C-NMR spectra indicate slight shift changes to lower field of the bridging CC carbon atoms. The bridging pyridine C-atoms are only shifted less than 1 ppm, so that the shift difference is within the measurement error. In the 1H-NMR spectra, however, the o-pyridine protons (Hα(py)) in complexes 4 and 5 are shifted upfield ca. 1.5 ppm, the m-pyridine protons (Hß(py) are shifted upfield ca. 0.2 ppm in comparison to compound 3. These NMR shift changes in the bridging 4-ethynylpyridine moiety might indicate a shift of electron density in this bridge towards the coordinated, positively charged Re ligand.
In the IR spectra, the asymmetric CC vibrations can be observed only as shoulders (2012 cm−1
(4), 2021 cm−1
(5)) due to their overlap with one of the carbonyl vibrations (2030 cm−1
(4), 2026 cm−1
(5)). The asymmetric stretching vibration is lowered ca. 50 cm−1 in both cases in comparison to the corresponding CC vibration of the precursor compound 3. The symmetric CC stretching vibration is found at 2048 cm−1
(4) and 2053 cm−1
(5), respectively, which are both lower values than that found for the symmetric CC stretching vibration of complex 3
(2070 cm−1). These changes are probably due to a weakening of the triple bond because of electron delocalization. The py(NC)-vibration is shifted to 1596 and 1599 cm−1, respectively, thus indicating a slight strengthening of the bond, possibly due to the increased electron density in the pyridine ring because of its bridging function.
The FAB-MS spectrum of compound 4 does not show a [M]+ peak, however, the [M–(Re(CO)3(bipy))]+ signal can be observed with a relative intensity of 20%. The signal of [M–2 (Re(CO)3(bipy))]+ occurs with an intensity of 8% and the signal of [Re(CO)3(bipy)]+ is the most intense peak of the spectrum (100%). As expected, the bridging N–Re bond is the easiest one to break under FAB conditions.
A symmetrical bipy and two intense and broad carbonyl stretching bands imply a facial disposition of the three carbonyl ligands about the rhenium centre in the dinuclear complexes 6 and 7, derived from compound 1 as the starting material according to eqn. (5). In both complexes, the hydride resonances appear as one high field quintet in their 1H NMR spectra (δ(1H)=−9.58 ppm, JPH=19.9 Hz in 6; δ(1H)=−9.58 ppm and JPH=20.1 Hz in 7). One singlet is observed in the 31P{1H} NMR spectra suggesting the equivalence of the P atoms in the ligand. Due to the phenyl, pyridine, and bipyridine moieties in the complexes 6 and 7 the 13C-NMR signals of the respective carbons are overlapping to a significant extent and prevent a detailed assignment (see experimental section) and interpretation. In the 1H-NMR spectra, as in the case of complexes 4 and 5 in comparison to compound 3
(see above), the o-pyridine protons (Hα(py)) in complexes 6 and 7 are shifted upfield ca. 1.5 ppm, the m-pyridine protons (Hß(py) are shifted upfield ca. 0.25 ppm in comparison to compound 1. The signals of the protons riding on the bridging carbons of the diphenyl ligands are, as in the case of compound 1 and 2 split due to the non-symmetric coordination situation above and below the Ru-diphosphine plane. The respective 1H NMR signals appear at 2.38 and 1.96 ppm (6) and 2.37 and 1.97 ppm (7), respectively. As can be seen in comparing these shifts to that observed in the spectrum of 1
(2.43/1.96), the influence of the Re ligand on them is negligible.
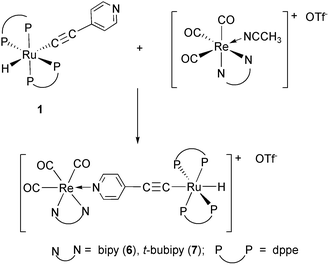 | (5) |
As observed for the asymmetric CC stretching vibration of the compounds 4 and 5, the CC-stretching vibration of the complexes 6 and 7 occurs as a shoulder (2022 and 2020 cm−1, respectively) of a carbonyl band and indicates a weakening of the triple bond (see above). In all complexes 4–7 in which the Ru(II) moiety is ligated by Re(carbonyl) complexes, the carbonyl stretching frequencies are generally found at slightly higher wavenumbers than in the not coordinated rhenium precursor complex [Re(N–N)(CO)3(MeCN)]OTf (N–N=bipy, t-bu2bipy, OTf=CF3SO3) and of the closely related Re(carbonyl) complex Br(CO)3Re(4,4′bipy)2.16 These observations reveal a decrease in the back-donation capability from the Re(I) center to the π* orbitals of the CO ligands.
Electrochemistry and spectroelectrochemistry of compounds 3 and [4]2+
Cyclic voltammograms of complexes 37a and [4]2+ as well as of the parent complex trans-Ru(dmpe)2Cl2 reveal an oxidation process (Fig. 1) at similar redox potentials (in the range +0.34 to +0.62 V) (Table 1). Accordingly, this process can be assigned to the Ru(II/III) oxidation. By comparison, the related complex trans-Ru(dppe)2(CCPh) has a similar oxidation process at E°=+0.52 V.17a,b The tiny signal at ca. 0.4 V in the cyclic voltammogram of compound [4]2+
(see Fig. 1) is likely to be caused by a small remnant of its precursor complex 3 in the sample of compound 4, since complex 3 has an oxidation wave at +0.42 V.
![Cyclic voltammograms of a solution of CH2Cl2, ButNPF6
(0.2 mol dm−3) and (a)
fac-[Re(CO)3(bipy)(py)]OTF (7.4 × 10−4 mol dm−3); (b)
3
(3.8 × 10−4 mol dm−3); (c)
[4](OTF)2
(5.0 × 10−4 mol dm−3). Scan rate 0.2 V s−1.](/image/article/2004/NJ/b307919h/b307919h-f1.gif) | ||
| Fig. 1 Cyclic voltammograms of a solution of CH2Cl2, ButNPF6
(0.2 mol dm−3) and (a)
fac-[Re(CO)3(bipy)(py)]OTF (7.4×10−4 mol dm−3); (b)
3
(3.8×10−4 mol dm−3); (c)
[4](OTF)2
(5.0×10−4 mol dm−3). Scan rate 0.2 Vs−1. | ||
| Complex | E°/Volt (ΔE/mV) | ||||
|---|---|---|---|---|---|
| a Irreversible process, affected by electrode absorption phenomena. b Irreversible process. c ECE process giving rise to a process at −1.20 V. d Difficult to distinguish due to solvent discharge. | |||||
| ReII/ReI | RuIV/RuIII | RuIII/RuII | bipy−/bipy | bipy2−/bipy− | |
| trans-Ru(dmpe)2Cl2 | — | +1.50a | +0.34 (70) | — | — |
| fac-[Re(CO)3(bipy)(py)]+ | +1.84b | — | — | −1.20 (60) | −1.7b |
| 3 | — | +1.40b | +0.42 (70) | — | — |
| [4]2+ | +1.84b | +1.48b | +0.62 (110) | −1.06bc | d |
A second, irreversible, oxidation process in the range +1.40 to +1.50 V is also visible in the cyclic voltammogram of all three complexes mentioned above. Since the oxidation of diphosphine is expected to have a much higher redox potential, this second oxidation process is tentatively ascribed to the irreversible Ru(III/IV) oxidation. Complex [4]2+ also undergoes a third, chemically irreversible, oxidation at +1.84 V. An irreversible reduction at −1.06 V is also present, followed by a further reversible reduction at −1.20 V. A comparison with the cyclic voltammogram of the complex fac-[Re(CO)3(bipy)(py)]+ helps assigning these further redox processes. Indeed, the third oxidation change of [4]2+ is found at the same potential as the Re(I/II) change in free fac-[Re(CO)3(bipy)(py)]+. This latter complex is presumably formed upon the irreversible oxidation of [4]3+ at +1.48 V. Furthermore, the reduction at −1.20 V is easily recognised as the redox change (bipy/bipy˙−) in free fac-[Re(CO)3(bipy)(py)]+, formed after the process at −1.06 V. Accordingly, it seems reasonable to ascribe the latter reduction to the equivalent redox change on the ligated fac-[Re(CO)3(bipy)(py)] units, subsequently followed by their disconnection. The cyclic voltammograms of compound 3, [4]2+ and fac-[Re(CO)3(bipy)(py)]+ are depicted in Fig. 1.
Bulk electrolysis (EW=+0.6 V) of complex 3 consumes more than 1 e− mol−1 and leads to decomposition of the electro-generated cation. At the same time the original pale yellow solution turns cherry-red. Similarly, bulk electrolysis (EW=+0.8 V) of complex [4]2+ consumes less than 1 e− mol−1 and leads to decomposition of the electro-generated trication, while the original pale yellow solution turns orange. In spite of the instability of the oxidised species, spectroelectrochemical analysis has been performed, it being possible to conduct such a study by very fast electrolysis in a tight OTTLE cell. As a general criterion, spectra have been collected until disappearance of the isosbestic points, interpreted as the sign of decomposition of the oxidised species. The wavelength values for 3, [4]2+, trans-Ru(dmpe)2Cl2 and their oxidised forms are summarised in Table 2, together with those of fac-[Re(CO)3(bipy)(py)]+ and its reduced form. The spectra shown in Fig. 2 reveal that the initial spectrum of complex [4]2+ is not a simple superposition of the spectra of the parent complexes 3 and fac-[Re(CO)3(bipy)(py)]+.
2(10.3 × 10−4 mol dm−3) and fac-[Re(CO)3(bipy)(py)]OTF (7.0 × 10−4 mol dm−3) in a solution of CH2Cl2 and ButNPF6
(0.2 mol dm−3).](/image/article/2004/NJ/b307919h/b307919h-f2.gif) | ||
| Fig. 2 OTTLE cell UV-vis-NIR spectra of 3
(4.6×10−4 mol dm−3), [4](OTF)2(10.3×10−4 mol dm−3) and fac-[Re(CO)3(bipy)(py)]OTF (7.0×10−4 mol dm−3) in a solution of CH2Cl2 and ButNPF6
(0.2 mol dm−3). | ||
| Complex | λ/nma | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a
sh=shoulder, w=weak, ↑ increasing on electrolysis, ↓ decreasing on electrolysis.
|
|||||||||||||||
| Ru(dmpe)2Cl2 | 242 | ||||||||||||||
| [Ru(dmpe)2Cl2]+ | 242↑ | 280s↑ | 369w↑ | 680w↑ | |||||||||||
| [Re(CO)3(bipy)(py)]+ | 238 | 266 | 302sh | 317 | 353w | ||||||||||
| Re(CO)3(bipy)(py) | 238 | 266↓ | 302sh | 317 | 353w | ||||||||||
| 3 | 247w | 354 | |||||||||||||
| [3]+ | 247w↓ | 288w↑ | 354↓ | 471w↑ | 893w↑ | ||||||||||
| [4]2+ | 237 | 281 | 300sh | 312sh | 354 | 418 | 446w | ||||||||
| [4]3+ | 235 | 281 | 300sh | 312sh | 353 | 397↑ | 418↓ | 868w↑ | |||||||
Indeed, after formation of complex [4]2+ from compound 3 and the rhenium ligand, a new band appears at 418 nm. The MLCT band (dπ(Ru)→π*(CCpy)) observed in 3 at λmax=354 nm is also still visible. As a consequence it is difficult to simply ascribe the signal at 418 nm to the bathochromic shift of the MLCT absorption, which, on the other hand, would be consistent with the Lewis acidity of the pyridine-bound Re(I) centers. The new band might be due to charge transfer between the metallic units in the trimetallic systems. For a heterometallic IT band belonging to Robin–Day class II, the bandwidth at half height (Δν1/2) is predicted to be given by the following formula,18
Δν1/2=[2130(![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) −ΔE°)]1/2 −ΔE°)]1/2 |
≈|2.8| V can be evaluated from the measured bandwidth (Δν1/2=1400 cm−1). Since ΔE°=E°(RuIII/RuII)−E°(ReI/Re0), the rhenium reduction process should be expected to take place at E°(ReI/Re0)≈−||2.8| V−0.62 V |=−2.2 V. Unfortunately, due to solvent discharge, we did not clearly observe this latter reduction process. Nevertheless, the calculated value seems very reasonable. Besides, redox processes lower than −2.0 V have been reported for [Re(CO)3(L)(py)]+ systems in MeCN (with L=bidentate aromatic N-donor), and attributed to reduction of the metal center.19 The OTTLE spectra of [4]2+ and its oxidation product are shown in Fig. 3b. On oxidation, the band at 418 nm rapidly diminishes in intensity and a new band appears at 397 nm, which is tentatively assigned to a LMCT transition. At the same time, the band at 354 remains almost unchanged on oxidation of [4]2+, while it diminishes on oxidation of the parent complex 3
(Fig. 3a). This behaviour is possibly due to incomplete oxidation of [4]2+. As previously stated, complete oxidation can not be achieved due to the decomposition of the electro-generated [4]3+. Finally, the appearance of two forbidden d–d transitions is also noticed on oxidation of 3
(at 471 and 893 nm), one of them being at very low energy. The low energy band is also found on oxidation of [4]2+, while the high energy band may be covered by stronger absorptions. These absorptions seem to be due to dπb–dπa transitions between the bonding and antibonding combination of dπ with π* orbitals of the ligand.
2(10.3 × 10−4 mol dm−3) in a solution of CH2Cl2 and ButNPF6
(0.2 mol dm−3). Spectra were recorded after subsequent 2 min intervals of applied potential (Ew = +0.4 V for 3 and +0.6 V for [4](OTF)2.](/image/article/2004/NJ/b307919h/b307919h-f3.gif) | ||
| Fig. 3 OTTLE cell Uv-vis-NIR spectra of (a)
3
(4.6×10−4 mol dm−3), (b)
[4](OTF)2(10.3×10−4 mol dm−3) in a solution of CH2Cl2 and ButNPF6
(0.2 mol dm−3). Spectra were recorded after subsequent 2 min intervals of applied potential (Ew=+0.4 V for 3 and +0.6 V for [4](OTF)2. | ||
Thermogravimetric examination of compounds 1–6
Surprisingly, the complexes 1–3 display a quite different decomposition behaviour. While compound 1 already starts decomposing at 127°C by losing its –CCpy ligand,14 the complex 2 is stable up to 317°C and complex 3 does not decompose before 418°C. Compound 1 is the only compound examined in this work which displays distinct decomposition steps, the first one starting at 127°C (mass loss ca. 10%), the second starting at 243°C. This second step, however lasts until the end of the measurement at 650°C where a mass loss of in total 57% is reached. The second decomposition step must be attributed to the partial degradation and loss of the dppe ligand. In the case of compound 2 the only observed decomposition step starts at 317°C, the mass loss until 650°C is about 27% of the original mass. The dppm ligand seems to stabilize the molecule much more than the dppe ligand. Even more pronounced is the stability of complex 3, which starts decomposing at 418°C, but then loses mass comparatively quickly. At 650°C only 16% of the original mass is left. The Re(I) compound [Re(2,2′-bipy)(CO)3(4,4′-bipy)]OTf,16d closely related to the Re-containing complexes 4–7 with regard to the ligand environment of the Re centre starts decomposing with the first decomposition onset at 202°C, due to the loss of the three carbonyls. The Re(I) precursor of compounds 4 and 6, [Re(bipy)(CO)3(NCCH3)]OTf, starts decomposing already below 100°C, due to partial loss of its weakly coordinated acetonitrile ligand.
In order to see several clearly distinct decomposition steps and the stabilizing influence of the Re(I) units the thermally less stable complex 1
(not the thermally more stable complex 2) was chosen as precursor for the complex 6. Both compounds 4 and 5 are significantly more stable than [Re(2,2′-bipy)(CO)3(4,4′-bipy)]OTf and their acetonitrile containing Re(I) precursor complexes, but less stable than the Ru(II) precursor 3. Compound 4 has the onset of its decomposition at ca. 371°C, about 170°C above the decomposition onset of [Re(2,2′-bipy)(CO)3(4,4′-bipy)]OTf. Until 650°C the mass loss, which again occurs only in one step without clearly pronounced features sums up to ca. 50%. The decomposition onset of compound 5 is observed at ca. 331°C. The degradation of this molecule is faster than that of compound 4 and results in a sublimation of all remnants of the decomposition (0% left at 650°C). Due to the high total mass of the complex, the salt character and the colour change during the decomposition a sublimation of the intact complex 5 can be excluded. Compound 6 starts decomposing at 369°C (end mass at 650°C: 58%). This is, with regard to the error range of the method, identical to the decomposition onset of complex 4. It might be assumed that at this temperature the Re(I) moieties break down, regardless of the nature of the attached molecule, since the Ru precursor complex 3 is somewhat more stable than complex 4, but complex 1 is significantly less stable than complex 6. Anyway, the stabilization of the Re(I) fragment in the compounds 4 and 6 in comparison to [Re(bipy)(CO)3(MeCN)]OTf (decomposition onset: 202°C) and the related complex Re(4,4′-bipy)2)(CO)3Br (decomposition onset: 222°C)16d is significant. The decomposition of the compounds 4–6 starts very likely with the breaking of the N(py)→Re bond, as indicated by MS experiments (see above).
X-ray structure analysis of [Re(CO)3(t-Bu2bpy)Ru(dppe)2(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) Cpy-4)H]OTf (7)
Cpy-4)H]OTf (7)
The structure of the heterobimetallic compound 7 was established by X-ray crystallography. Fig. 4 illustrates the distorted octahedral ligand environment of both the Ru and the Re atom and the bridging 4-ethynylpyridine moiety. Selected bond angles and bond distances are listed in Table 3. The hydride atom bonded to the ruthenium was found in the difference Fourier map and refined. To the best of our knowledge, among the few ruthenium hydride alkynyl complexes known to date, it was possible in only two cases to refine the hydride. Our results in the case of complex 7 confirm very well these earlier findings. The Ru–H bond distances (165(5) pm in the case of complex 7, 161(5) pm in the case of HRu(dmpe)2–CCpy–Rh2(Ac4)–pyCC–Ru(dmpe)2H, 157(8) pm in [HRu(CCPh)(PPh3)3]·(C6H6) are identical within the error range of the measurement.7a,15c,d Based on these three results it seems that the unrefined value of 135 pm for the Ru–H distance in [Cp*RuH(CC–COOMe)(dppe)][BPh4]15c,d is probably too short.
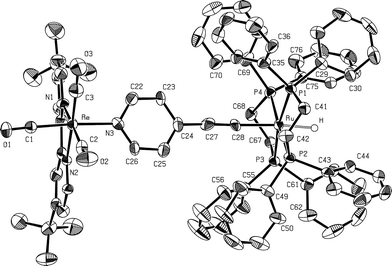 | ||
| Fig. 4 ORTEP28 style drawing of the solid state structure of compound 7. The thermal ellipsoids are given at a 50% probability level. | ||
| Re–N1 | 215.6(8) | Ru–C28 | 200.4(5) |
| Re–N2 | 219.2(5) | Ru–H | 165(5) |
| Re–N3 | 218.1(5) | Ru–P1 | 234.96(16) |
| Re–C1 | 192.5(5) | Ru–P2 | 232.27(17) |
| Re–C2 | 197.3(9) | Ru–P3 | 233.09(16) |
| Re–C3 | 185.1(9) | Ru–P4 | 233.06(18) |
| C27–C28 | 129.8(9) | P1–Ru–P2 | 83.62(6) |
| N1–Re–N2 | 73.6(2) | P3–Ru–P4 | 83.34(6) |
| N1–Re–N3 | 83.8(2) | C28–Ru–H | 173.7(19) |
| N2–Re–N3 | 84.1(2) | P1–Ru–H | 86.6(18) |
| N3–Re–C1 | 172.9(3) | P2–Ru–H | 82.1(19) |
| N3–Re–C2 | 93.6(2) | P3–Ru–H | 84.3(18) |
| N3–Re–C3 | 94.9(3) | P4–Ru–H | 94.6(19) |
| C24–C27–C28 | 177.1(8) | Ru–C28–C27 | 177.3(6) |
The Ru–C28 distance (200.4(5) pm) is slightly shorter than that reported for HRu(dmpe)2–CCpy–Rh2(Ac4)–pyCC–Ru(dmpe)2H (207.1(2) pm)7a and that observed for another related complex containing the [(CO)3Re(bipy)–pyCC–Ru(16-TMC)–CCpyRe(bipy)(CO)3]2+ cation (16-TMC=1,5,9,13-tetramethyl-1,5,9,13-tetraazacyclohexadecane)
(204.5(10) pm).4e The bond distance of the bridging C27–C28 atoms (129.8(9) pm), however, is somewhat longer than in the two aforementioned compounds (119.7(3) and 120.7(13) pm, respectively).4e,7a The observation of a relative long CC bond distance in compound 7 is in accord with the observations made in the vibrational spectra, indicating a weakening of this bond in comparison to the precursor molecule 1
(see above). The Re–N3 bond distance is 218.1(5) pm, similar to that in [(CO)3t-bu2bipy)Re–pyCC–Ru(form)2]2
(220.7(3) pm),16e and in [(CO)3Re(bipy)–pyCC–Ru(16-TMC)–CCpyRe(bipy)(CO)3]2+
(220.8(7) pm).4e
Conclusions
Several new Ru(II) σ-alkynyl pyridine complexes containing conjugated pyridine ligands have been prepared. All described complexes are very stable both in the solid state and in solution and useful in the construction of multi-nuclear complexes.In the linear complexes resulting from the reaction of Ru(II) hydride alkynyl pyridine moieties with [Re(I)]+ building blocks the ruthenium alkynyl complexes behave as electron donors. Related complexes might be useful precursors of nonlinear optical materials. Using the less bulky phosphine ligand dmpe, an organometallic linear-rod building block, Ru(dmpe)2(CCpy-4)2 can be prepared. It is also a very versatile moiety for constructing heterobimetallic units. Spectroscopic, thermal and electronic investigations indicate that significant stabilizing interactions exist between the ruthenium and the attached rhenium units.
Based on the findings presented here it should be possible to synthesize stable molecular rods based on Ru units ligated with two σ-alkynyl pyridine ligands. Work in this direction is currently under way in our laboratory.
Experimental section
Unless stated otherwise, all reactions and manipulations were carried out in dry glassware under nitrogen or argon atmosphere. Solvents were dried by standard procedures. 4-Ethynylpyridine,20trans-Ru(dppe)2Cl2, cis-Ru(dppe)2Cl2,21cis-Ru(dppm)2Cl2,22 [Re(N–N)(CO)3(MeCN)]OTf (N–N=bipy, t-bu2bipy, OTf=CF3SO3)8a were prepared according to the literature methods. Elemental analyses were performed in the Mikroanalytisches Labor of the TU München in Garching (Mr M. Barth). 1H, 13C and 31P{1H} NMR were obtained with a Bruker Avance DPX-400 spectrometer. IR (in KBr pellets) and Raman spectra (in powder samples) were obtained on a Bio-Rad FTS-575C spectrometer at room temperature. Electronic absorption spectra were run using a PERKIN-ELMER Lambda 2 UV/VIS spectrometer. Mass spectra were obtained with Finnigan MAT 311 A and MAT 90 spectrometers. Electrochemical measurements were performed in a dichloromethane solution containing ButNPF6
(2.0×10−3 mol dm−3) as supporting electrolyte. Anhydrous 99.9% dichloromethane was obtained from Aldrich. Electrochemical grade ButNPF6 was purchased from Fluka and used as obtained. Cyclic voltammetry was performed in a three-electrode cell containing a platinum working electrode surrounded by a platinum-spiral counter electrode, and an aqueous saturated calomel reference electrode (SCE) mounted with a Luggin capillary. A BAS 100 W electrochemical analyser was used as polarising unit. All the potential values are referred to the saturated calomel electrode (SCE). Under the present experimental conditions, the one-electron oxidation of ferrocene occurs at E°′=+0.39 V. Controlled potential coulometry was performed in an H-shaped cell with anodic and cathodic compartments separated by a sintered-glass disk. The working macroelectrode was a platinum gauze; a mercury pool was used as the counter electrode. The UV-vis spectroelectrochemical measurements were carried out using a Perkin-Elmer Lambda 900 UV-vis spectrophotometer and an OTTLE (optically transparent thin-layer electrode) cell23 equipped with a Pt-minigrid working electrode (32 wires cm−1), Pt minigrid auxiliary electrode, Ag wire pseudo reference and CaF2 windows. The electrode potential was controlled during electrolysis by an Amel potentiostat 2059 equipped with an Amel function generator 568. Nitrogen-saturated dichloromethane solutions of the compound under study were used with ButNPF6
(2.0×10−3 mol dm−3) as supporting electrolyte.
[trans-Ru(dppe)2H(CCpy-4)]
(1)
4-Ethynylpyridine (103.0 mg, 1.0 mmol) was added to a solution of cis-Ru(dppe)2Cl2
(290.6 mg, 0.30 mmol) in methanol (20 ml). The solution was stirred for 10 min and then sodium (130 mg) was added. The mixture was refluxed for 5 h. It was then cooled to room temperature and the resulting solid was filtered off and washed with methanol and Et2O. The solid was extracted with 20 ml CHCl3 and then the solvent was removed under vacuum. The remaining pale yellow solid was washed with Et2O and dried in vacuo. The product was further purified by recrystallization from CHCl3/Et2O. Yield: 190 mg (63%). Anal. Calc. for C59H53NP4Ru (1001.05): C, 70.79; H, 5.33; N, 1.40%. Found, C, 70.45; H, 5.40; N, 1.15%. IR (KBr, cm−1): 3050 (m), 2964 (w), 2055 (s), 1823 (w), 1584 (s), 1433 (s), 1093 (m), 821 (m), 741 (m), 693 (s), 531 (s), 490 (m), 427 (w). 1H NMR (CDCl3): δ 8.31 (d, JH–H=5.2 Hz, 2H, Hα), 6.92–7.42 (m, 40H, Ph), 6.82 (d, JH–H=5.2 Hz, 2H, Hβ), 2.43 (m, 4H, CH2), 1.96 (m, 4H, CH2), −9.71 (qn, JP–H=22.0 Hz, 1H, Ru–H). 13C NMR (CDCl3): δ 149.2 (m, Ru–C), 148.3 (Cα(py)), 139.3/138.8 (dt (Cα-phenyl)), 136.6 (Cγ(py)), 133.0/132.8 (dt, Cβ-phenyl), 128.8/128.7 (d, Cδ-phenyl), 128.5/128.4 (dt, Cγ-phenyl), 124.9 (Cβ(py)), 110.0 (s, Ru–CC), 33.4 (m, P–CH2). 31P{1H} NMR (CDCl3): δ 67.7 (s). FAB-MS (Nitrobenzyl alcohol as FAB matrix, Ru 102), m/z=1001 ([M]+, rel. int. 20%), 899 ([Ru(dppe)2H]+, 100%).
[trans-Ru(dppm)2H(CCpy-4)]
(2)
4-Ethynylpyridine (103.0 mg, 1.0 mmol) was added to a solution of cis-Ru(dppm)2Cl2
(282.0 mg, 0.30 mmol) in methanol (25 ml). The solution was stirred for 10 min and sodium (130 mg) was added. The mixture was refluxed for 5 h and it was then cooled to room temperature. The resulting solid was filtered off and washed with 3×10 ml n-hexane. The solid was extracted with 20 ml CHCl3 and then the solvent was removed under vacuum. The pale yellow solid was washed with n-hexane and dried in vacuo. Yield: 175 mg (60%). Anal. Calc. for C57H49NP4Ru (972.99): C, 70.36; H, 5.08; N, 1.44%. Found, C, 70.55; H, 5.34; N, 1.25%. IR (KBr, cm−1): 3050 (m), 2962 (w), 2066 (s), 1772 (w), 1584 (s), 1434 (s), 1096 (m), 727 (s), 693 (s), 505 (s), 482 (m), 429 (w). 1H NMR (CDCl3): δ 8.09 (d, JH–H=6.1 Hz, 2H, Hα), 7.06–7.44 (m, 40H, Ph), 6.27 (d, JH–H=6.1 Hz, Hβ), 4.78 (m, 2H, CH2), 4.53 (m, 2H, CH2), −6.30 (qn, JPH=19.9 Hz, 1H, Ru–H). 13C NMR (CDCl3): δ 148.3 (Cα(py)), 148.0 (Ru–C), 139.7/136.7 (dt, (Cα-phenyl)), 136.7 (Cγ(py)), 133.0/132.7 (dt, Cβ-phenyl), 129.3/129.1 (d, Cδ-phenyl), 127.9 and 127.8 (d, Cγ-phenyl), 124.9 (Cβ(py)), 117.7 (Ru–CC), 56.1 (t, P–CH2). 31P{1H} NMR (CDCl3): δ 2.3 (s). FAB-MS (Nitrobenzyl alcohol as FAB matrix, Ru 102), m/z=973 ([M]+, rel. int. 12%), 871 ([Ru(dppm)2H]+, 100%).
[trans-Ru(dmpe)2(CCpy-4)2]
(3)
A solution of trans-Ru(dmpe)2Cl2
(100 mg, 0.21 mmol) in 2-propanol/THF (10∶90, 25 ml) was stirred for 10 min at room temperature. Sodium (220 mg) was added and the solution was stirred for 30 h. The solvent was removed in vacuo and the residue was extracted with n-pentane. The extracts were evaporated to dryness to afford [Ru(dmpe)2H2] and used without further purification. It was dissolved in MeOH (15 ml) and 4-ethynylpyridine (127 mg, 1.2 mmol) was added. The mixture was refluxed for 1.5 h and subsequently stirred overnight at room temperature. The solution was concentrated to 5 ml. The pale yellow solid was filtered off, washed with MeOH and Et2O and dried in vacuo. Yield: 89.0 mg (70%). Anal. Calc. for C26H40N2P4Ru (605.58): C, 51.57; H, 6.66; N, 4.63%. Found, C, 51.13; H, 6.59; N, 4.32%. IR (KBr, cm−1), 3054 (w), 2965 (w), 2900 (m), 2070 (s), 2059 (s) 1583 (s), 1449 (s, br), 1204 (m), 941 (m), 839 (m), 817 (m), 710 (m), 647 (m), 528 (m). λmax(CH2Cl2): 249.0 (logε 4.16), 358 nm (4.54). 1H NMR (CD2Cl2,): δ 8.16 (d, JH–H=5.9 Hz, 4H, Hα), 6.81 (d, JH–H=6.0 Hz, 4H, Hβ), 1.71 (t, JH–H=9.0 Hz, 8H, P–CH2), 1.55 (s, 24H, P–CH3). 13C NMR (CD2Cl2): δ 149.7 (Cα(py)), 144.6 (Ru–C), 138.6 (Cγ(py)), 125.5 (Cβ(py)), 109.5 (Ru–CC), 30.8 (m, P–CH2), 16.2 (m, P–CH3). 31P{1H} NMR (CD2Cl2), δ 38.9 (s). FAB-MS (Nitrobenzyl alcohol as FAB matrix, Ru 102), m/z=606 ([M]+, rel. int. 18%), 504 ([Ru(dppm)2(CCpy)]+, 28%), 402 ([Ru(dppm)2]+, 31%).
[Re(CO)3(bipy)]2[trans-Ru(dmpe)2(CCpy-4)2](OTf)2(4)
A solution of [Re(bipy)(CO)3(MeCN)]OTf (129.5 mg, 0.21 mmol) and 3
(60.5 mg, 0.10 mmol) in THF (20 ml) was refluxed in the dark for 5 h. It was cooled to room temperature and the resulting solid was filtered off to yield the products as yellow powder (123 mg, 70%). The orange crystals were obtained after the recrystallization using acetonitrile and Et2O. Anal. Calcd. for C54H56N6F6O12P4S2Re2Ru (1756.57): C, 36.92; H, 3.21; N, 4.78%. Found: C, 37.02; H, 3.20; N, 4.71%. IR (cm−1, KBr), 2964 (w), 2904(w), 2048 (m) 2030 (vs), 2012 (s), 1920(vs), 1596(s), 1265 (s, br), 1151 (m), 1032 (s), 802(m), 637 (m), 517 (w). λmax(CH2Cl2): 292.0 (logε 4.39), 423 nm (4.62). 1H NMR (CD3CN): δ 9.14 (d, JH–H=4.5 Hz, 4H, bipy), 8.35 (d, JH–H=8.2 Hz, 4H, bipy), 8.21 (m, 4H, bipy), 7.73 (m, 4H, bipy), 7.67 (d, JH–H=6.8 Hz, 4H, Hα), 6.59 (d, JH–H=6.7 Hz, 4H, Hβ), 1.59 (t, 8H, P–CH2), 1.36 (s, 24H, P–CH3). 13C NMR (CD3CN): δ 196.6, 192.0 (CO), 154.6 (C(bipy)(bridging)), 150.2 (Cα(bipy)), 149.5 (Cα(py)), 147.9 (Ru–C), 139.5(Cγ(py)), 141.9 (Cγ(bipy)), 127.6, 124.1 (Cβ(bipy)), 125.1 (Cβ(py)), 111.5 (CC–Ru), 30.3 (P–CH2), 15.4 (P–CH3). 31P{1H} NMR (CD3CN), δ 37.7 (s). FAB-MS (Nitrobenzyl alcohol as FAB matrix, Ru 102, Re 187), m/z=1033 ([M–Re(CO)3(bipy)]+, rel. int. 24%), 606 ([Ru(dmpe)2(CCpy)]+, 8%), 427 ([Re(CO)3(bipy)]+, 100%).
[Re(CO)3(t-bu2bipy)]2[trans-Ru(dmpe)2(CCpy-4)2](OTf)2
(5)
A solution of [Re(t-bu2bipy)(CO)3(MeCN)]OTf (153 mg, 0.21 mmol) and 3
(60.5 mg, 0.10 mmol) in THF (20 ml) was refluxed in the dark for 4 h. The solvent was then reduced to 5 ml under vacuum and 20 ml Et2O was added to precipitate the solid. After filtering, the yellow solid was washed with 3×5 ml Et2O, dried in vacuo. The recrystallization of the crude product in CH2Cl2/Et2O afforded 79.2 mg (40% yield) of 5 as yellow crystals. Anal. Calcd. for C70H88N6F6O12P4S2Re2Ru (1981.0): C, 42.44; H, 4.48; N, 4.24%. Found: C, 42.02; H, 4.33; N, 4.03%. IR (cm−1, KBr), 2966 (w), 2907 (w), 2053 (m) 2026 (vs), 2021 (s), 1916 (vs), 1599 (s), 1261 (m), 1031 (m), 843 (m), 638 (m), 557 (w). λmax(CH2Cl2): 282.0 (logε 4.47), 421.0 nm (4.65). 1H NMR (CD2Cl2): δ 8.95 (d, JH–H=6.0 Hz, 4H, t-bu2bipy), 8.23 (d, JH–H=2.2 Hz, 4H, t-bu2bipy), 7.68 (dd, JH–H=2.2, 6.0 Hz, 4H, t-bu2bipy), 7.65 (d, JH–H=6.8 Hz, 4H, Hα), 6.65 (d, JH–H=6.8 Hz, 4H, Hβ), 1.61 (t, 8H, P–CH2), 1.44 (s, 18H, C(CH3)3), 1.39 (s, 24H, P–CH3). 13C NMR (CD2Cl2): δ 195.5, 191.1 (CO), 165.5 (Cγ(t-bu2bipy)), 155.0(C (t-bu2bipy)(bridging)), 152.3 (Cα(t-bu2bipy)(CH)), 149.1 (Cα(py)), 147.9 (Ru–C), 139.5 (Cγ(py)), 125.1 (Cβ(py)), 123.7/120.0 (Cβ(t-bu2bipy)), 111.0 (CC–Ru), 35.0 (CMe3), 29.3 (CH3), 29.2 (m, P–CH2), 14.4 (P–CH3). 31P{1H} NMR (CD2Cl2): δ 37.6 (s).
[Re(CO)3(bipy)Ru(dppe)2(CCpy-4)H]OTf (6)
A solution of [Re(bipy)(CO)3(MeCN)]OTf (61.6 mg, 0.10 mmol) and 1
(100.1 mg, 0.10 mmol) in THF (20 ml) was refluxed in the dark for 5 h. Evaporated the solvent under vacuum and the yellow solid was washed with 3 ml MeOH and 3×5 ml Et2O, then dried in vacuo. Orange needle crystals of 6 can be obtained after recrystallization from CH2Cl2/Et2O in 70% yield (110 mg). Anal. Calcd. for C73H61N3F3O6P4SReRu·CH2Cl2
(1673.49): C, 53.5; H, 3.82; N, 2.53%. Found: C, 53.14; H, 3.99; N, 2.76%. IR (cm−1, KBr), 3052 (w), 2924 (w), 2030 (vs), 2022 (sh), 1926 (vs), 1601 (s), 1433 (m), 1259 (m, br), 1029 (m), 697 (m), 637 (m), 531 (m). 1H NMR (CD2Cl2): δ 9.16 (d, 2H, bipy), 8.70 (d, 2H, bipy), 8.38 (d, 2H, bipy), 7.81 (m, 2H, bipy), 7.65 (d, 2H, Hα), 6.54 (d, 2H, Hβ), 6.91–7.39 (m, 40H, phenyl), 2.38 (t, 4H, P–CH2), 1.96 (t, 4H, P–CH2), −9.58 (qn, JP–H=19.9 Hz, 1H). 13C NMR (CD2Cl2): δ 197.0, 193.2 (CO), 156.5–149.7 (m, Cα(bipy, bridging), Ru–C, Cα(bipy), Cα(py)), 141.7–138.3 (m, Cγ(py), Cγ(bipy), Cα-phenyl), 133.7–124.7 (Cβ-phenyl, Cδ-phenyl, Cγ-phenyl, Cβ(py), Cβ(bipy)), 113.3 (Ru–CC), 33.3 (m, P–CH2). 31P{1H} NMR (CD2Cl2), δ 67.0 (s).
[Re(CO)3(t-bu2bipy)Ru(dppe)2(CCpy-4)H]OTf (7)
A solution of [Re(t-bu2bipy)(CO)3(MeCN)]OTf (72.9 mg, 0.10 mmol) and 1
(100.1 mg, 0.10 mmol) in THF (20 ml) was refluxed in the dark for 3 h. Evaporated the solvent under vacuum and the yellow solid obtained was washed with 3×5 ml Et2O, then dried in vacuo. After re-crystallization from CH2Cl2/Et2O, yellow plate crystals of 7 can be obtained in 70% yield (120 mg). Anal. Calcd. for C81H77N3F3O6P4SReRu (1688.69): C, 57.61; H, 4.60; N, 2.49%. Found: C, 57.21; H, 4.47; N, 2.11%. IR (cm−1, KBr), 3051 (w), 2965 (w), 2025 (vs), 2020 (sh), 1924 (vs), 1601 (s), 1264 (m, br), 1031 (m), 696 (m), 637 (m), 531 (m). λmax(CH2Cl2): 315.0 (logε 4.16), 409.0 nm (4.34). 1H NMR (CD2Cl2): δ 9.09 (d, 2H, t-bu2bipy), 8.17 (d, 2H, t-bu2bipy), 7.80 (m, 2H, t-bu2bipy), 7.67 (d, 2H, Hα(py)), 6.59 (d, 2H, Hβ(py)), 6.91–7.37 (m, 40H, Ph), 2.37 (t, 4H, P–CH2), 1.97 (t, 4H, P–CH2), 1.47 (s, 18H, C(CH3)3), −9.58 (qn, JP–H=20.1 Hz, 1H). 13C NMR (CD2Cl2): δ 197.2, 192.9 (CO), 165.0 Cγ(t-bu2bipy), 155.5–148.5 (m, Cα(t-bu2bipy, bridging), Ru–C, Cα(bipy), Cα(py)), 142.1–139.1 (m, Cγ(py), Cα-phenyl), 131.9–121.4 (Cβ-phenyl, Cδ-phenyl, Cγ-phenyl, Cβ(py), Cβ(t-bu2bipy)), 115.1 (Ru–CC), 36.4 (CMe3), 30.3 (m, P–CH2), 29.5 (CH3). 31P{1H} NMR (CD2Cl2): δ 66.9 (s).
X-ray diffraction studies
Crystal data: C80H77N3O3P4ReRu, CF3O3S, Mr=1688.69, yellow crystal, 0.05×0.51×0.51 mm3, monoclinic, space group Pn
(No. 7), a=1335.30(1), b=1757.79(1), c=1749.87(2) pm, β=99.3274(3)°, V=4052.95(6)·106 pm3, Z=2, ρcalcd=1.384 g cm−3, F(000)=1708, μ(Mo-Kα)=1.841 mm−1, T=173 K. Data collection: A crystal of compound 7 suitable for diffraction experiments was selected in perfluorinated ether, and fixed in a capillary. Preliminary examination and data collection were carried out on a Nonius KappaCCD device at the window of a rotating anode X-ray generator and graphite monochromated Mo-Kα radiation (λ=71.073 pm), controlled by the COLLECT software package.24 911 collected images were processed using Denzo. Absorption and/or decay effects were corrected during the scaling procedure.25 A total of 74606 reflections were integrated and scaled. After merging (Rint=0.061) 14055 [13234: Io>2σ(Io)] independent reflections remained and all were used in the subsequent refinements. Structure solution and refinement: The structures were solved by direct methods26 and refined with standard difference Fourier techniques.27 Three non-hydrogen atoms (N3, C1, C3) located near the rhenium atom had to be refined isotropically. The necessity for this isotropical refinement is caused by an imperfect absorption correction (the crystal under examination was a very thin plate with irregular shape). All other non-hydrogen atoms of the asymmetric unit were refined with anisotropic thermal displacement parameters. All hydrogen atoms were calculated in ideal positions (riding model). The hydride atom bound to the ruthenium atom was found in the difference Fourier maps and refined freely with an individual isotropic thermal displacement parameter. Full-matrix least-squares refinements were carried out by minimizing ∑w(F2o−F2c)2 with SHELXL-97 weighting scheme and stopped at maximum shift/err <0.001. As can be seen by Flack's parameter ε=0.515(4) the crystal is twinned. The final model (including twin refinement, 897 parameters) converged with R1=0.0396 [Io>2σ(Io)], wR2=0.1040 [all data], and GOF=1.031. CCDC reference number 215603 (7). See http://www.rsc.org/suppdata/nj/b3/b307919h/ for crystallographic data in .cif or other electronic format.
Acknowledgements
The FCI is acknowledged for financial support of FEK. JLZ thanks the Alexander von Humboldt Foundation for postdoctoral research fellowships and Dr S. Zhen for experimental assistance. AMS is grateful to the FCT and the Alexander von Humboldt Foundation for a postdoctoral research fellowship and JZ thanks the DAAD for a PhD stipend.References
- (a) J. M. Lehn, Supramolecular Chemistry: Concepts and Perspective, VCH, Weinheim, 1995, ch. 8 Search PubMed; (b) Molecular Electronics, eds. J. Jortner and M. A. Ratner, Blackwell, Cambridge, 1997 Search PubMed.
-
(a) J. Michl, Science, 1991, 252, 511;
(b) H. Zhao, R. A. Heintz, K. R. Dunbar and R. D. Rogers, J. Am. Chem. Soc., 1996, 118, 12844 CAS;
(c) Y. Zhu, O. Clot, M. O. Wolf and M. G. P. A. Yap, J. Am. Chem. Soc., 1998, 120, 1812 CrossRef CAS;
(d) P. F. A. Schwab, M. D. Levin and J. Michl, Chem. Rev., 1999, 99, 1863 CrossRef CAS;
(e) R. P. Kingsborough and T. M. Swager, Prog. Inorg. Chem., 1999, 48, 123 CAS;
(f) T. Ren, G. Zou and J. C. Alvarez, Chem. Commun., 2000, 1197 RSC;
(g) K. T. Wong, J. M. Lehn, S. M. Peng and G. H. Lee, Chem. Commun., 2000, 2259 RSC;
(h) A. El-Ghayoury, A. Harriman, A. Khatyr and R. Ziessel, Angew. Chem. Int. Ed., 2000, 39, 185 CrossRef CAS;
(i) M. Abrantes, A. A. Valente, M. Pillinger, I. S. Gonçalves, J. Rocha and C. C. Romão, J. Catal., 2002, 209, 237 CrossRef CAS;
(j) M. Abrantes, A. A. Valente, M. Pillinger, I. S. Gonçalves, J. Rocha and C. C. Romão, Chem. Eur. J., 2003, 9, 2685 CrossRef CAS.
- (a) Extended Linear Chain Compounds, ed. J. S. Miller, Plenum, New York, 1982 Search PubMed; (b) N. G. Connelly and W. E. Geiger, Adv. Organomet. Chem., 1985, 24, 87; (c) S. R. Marder, Inorganic Materials, eds. D. W. Bruce and D. O'Hare, Wiley, Chichester, 1992, p. 115 Search PubMed; (d) F. A. Cotton, L. M. Daniels, C. Lin and C. A. Murillo, J. Am. Chem. Soc., 1999, 121, 4538 CrossRef CAS; (e) M. Chisholm, Acc. Chem. Res., 2000, 33, 53 CrossRef CAS; (f) F. A. Cotton, C. Lin and C. A. Murillo, Chem. Commun., 2001, 11 RSC; (g) W. M. Xue, F. E. Kühn, E. Herdtweck and Q. Li, Eur. J. Inorg. Chem., 2001, 213 CrossRef CAS; (h) F. A. Cotton, C. Lin and C. A. Murillo, Acc. Chem. Res., 2001, 34, 759 CrossRef CAS; (i) S. K. Hurst and T. Ren, J. Organomet. Chem., 2003, 670, 188 CrossRef CAS.
- (a) J. C. Calabrese, L. T. Cheng, J. C. Green, S. R. Marder and W. Tam, J. Am. Chem. Soc., 1991, 113, 7227 CrossRef CAS; (b) N. J. Long, Angew. Chem., Int. Ed. Engl., 1995, 34, 21 CrossRef CAS; (c) I. Y. Wu, J. T. Lin, J. Luo, S. S. Sun, C. S. Li, K. J. Lin, C. Tsai, C. C. Hsu and J. L. Lin, Organometallics, 1997, 16, 2038 CrossRef CAS; (d) I. R. Whittall, A. M. McDonagh, M. G. Humphrey and M. Samoc, Adv. Organomet. Chem., 1998, 42, 291; (e) M. Y. Choi, M. C. W. Chan, S. M. Peng, K. K. Cheung and C. M. Che, Chem. Commun., 2000, 1259 RSC.
-
(a) D. R. Kanis, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 1992, 114, 10338 CAS;
(b) J. Manna, K. D. John and M. D. Hopkins, Adv. Organomet. Chem., 1995, 38, 79 CAS;
(c) O. Lavastre, J. Plass, P. Bachmann, S. Guesmi, C. Moinet and P. H. Dixneuf, Organometallics, 1997, 16, 184 CrossRef CAS;
(d) A. M. McDonagh, M. G. Humphrey, M. Samoc, B. Luther-Davies, S. Houbrechts, T. Wada, H. Sasabe and A. Persoons, J. Am. Chem. Soc., 1999, 121, 1405 CrossRef CAS.
- (a) L. Dahlenburg, K. M. Frosin, S. Kerstan and D. Werner, J. Organomet. Chem., 1991, 407, 115 CrossRef CAS; (b) C. Bianchini, M. Peruzzini, F. Zanobini, P. Frediani and A. Albinati, J. Am. Chem. Soc., 1991, 113, 5453 CrossRef CAS; (c) M. I. Bruce, D. N. Duffy, M. J. Liddell, E. R. T. Tiekink and B. Nicholson, Organometallics, 1992, 11, 1527 CrossRef CAS; (d) W. Beck, B. Niemer and M. Wieser, Angew. Chem. Int. Ed. Engl., 1993, 32, 923 CrossRef; (e) L. S. Hegedus, Organometallics in Synthesis, ed. M. Schlosser, Wiley, New York, 1994, p. 383 Search PubMed.
- (a) J. L. Zuo, E. Herdtweck, F. Fabrizi de Biani, A. M. Santos and F. E. Kühn, New J. Chem., 2002, 26, 889 RSC; (b) J. L. Zuo, F. Fabrizi de Biani, A. M. Santos, K. Köhler and F. E. Kühn, Eur. J. Inorg. Chem., 2003, 449 CrossRef CAS.
- (a) J. K. Hino, L. Della Ciana, W. J. Dressick and P. Sullivan, Inorg. Chem., 1992, 31, 1072 CrossRef CAS; (b) W. M. Xue, M. C. W. Chan, Z. M. Su, K. K. Cheung, S. T. Liu and C. M. Che, Organometallics, 1998, 17, 1622 CrossRef CAS; (c) C. G. Garcia, J. F. de Lima and N. Y. M. Iha, Coord. Chem. Rev., 2000, 196, 219 CrossRef CAS; (d) H. Masui, Coord. Chem. Rev., 2001, 219, 957 CrossRef; (e) S. S. Sun and A. J. Lees, Organometallics, 2002, 21, 39 CrossRef CAS; (f) E. J. Schutte and B. P. Sullivan, Inorg. Synth., 2002, 33, 227 CAS.
- (a) E. Rojas, A. Santos, V. Moreno and C. Del Pino, J. Organomet. Chem., 1979, 181, 365 CrossRef CAS; (b) R. Nast and A. Beyer, J. Organomet. Chem., 1981, 204, 267 CrossRef CAS; (c) A. Romero, D. Peron and P. H. Dixneuf, J. Chem. Soc., Chem. Commun., 1990, 1410 RSC; (d) Y. Sun, N. J. Taylor and A. J. Carty, Organometallics, 1992, 11, 4293 CrossRef CAS.
- (a) M. I. Bruce and A. G. Swincer, Adv. Organomet. Chem., 1983, 22, 59 CAS; (b) D. Touchard, P. Haquette, N. Pirio, L. Toupet and P. H. Dixneuf, Organometallics, 1993, 12, 3132 CrossRef CAS.
- (a) P. Chow, D. Zargarian, N. J. Taylor and T. B. Marder, J. Chem. Soc., Chem. Commun., 1989, 1545 RSC; (b) P. J. Stang and C. M. Crittell, Organometallics, 1990, 9, 3191 CrossRef CAS.
- A. Kaplan and R. G. Bergman, Organometallics, 1998, 17, 5072 CrossRef CAS.
- Z. Atherton, C. W. Faulkner, S. l. Ingham, A. K. Kakkar, M. S. Khan, J. Lewis, N. J. Long and P. R. Raithby, J. Organomet. Chem., 1993, 462, 265 CrossRef CAS.
- (a) L. O. Field, A. V. George, D. C. R. Hockless, G. R. Purches and A. H. White, J. Chem. Soc., Dalton Trans., 1996, 2011 RSC; (b) M. Y. Choi, M. C. W. Chan, S. Zhang, K. K. Cheung, C. M. Che and K. Y. Wong, Organometallics, 1999, 18, 2074 CrossRef CAS.
- (a) J. Silvestre and R. Hoffmann, Helv. Chim. Acta, 1985, 68, 1461 CrossRef CAS; (b) M. I. Bruce, Chem. Rev., 1991, 91, 197 CrossRef CAS and references therein; (c) C. Bianchini, P. Frediani, D. Masi, M. Peruzzini and F. Zanobini, Organometallics, 1994, 13, 4616 CrossRef CAS; (d) I. de los Ríos, M. J. Tenorio, M. C. Puerta and P. Valerga, J. Am. Chem. Soc., 1997, 119, 6529 CrossRef CAS; (e) C. Bruneau and P. H. Dixneuf, Acc. Chem. Res., 1999, 32, 311 CrossRef CAS.
- (a) W. M. Xue, F. E. Kühn, G. Zhang, E. Herdtweck and G. Raudaschl-Sieber, J. Chem. Soc., Dalton Trans., 1999, 4103 RSC; (b) W. M. Xue, F. E. Kühn, G. Zhang and E. Herdtweck, J. Organomet. Chem., 2000, 596, 177 CrossRef CAS; (c) W. M. Xue and F. E. Kühn, Eur. J. Inorg. Chem., 2001, 2041 CrossRef CAS; (d) W. M. Xue, F. E. Kühn and E. Herdtweck, Polyhedron, 2001, 20, 791 CrossRef CAS; (e) J. L. Zuo, E. Herdtweck and F. E. Kühn, J. Chem. Soc., Dalton Trans., 2002, 1244 RSC.
- (a) C. E. Powell, M. P. Cifuentes, J. P. Morrall, R. Stranger, M. G. Humphrey, M. Samoc, B. Luther-Davies and G. A. Heath, J. Am. Chem. Soc., 2003, 125, 602 CrossRef CAS; (b) The reported redox potential of +0.60 V vs. Ag/AgCl has been converted to be referenced to S.C.E.
- (a) N. S. Hush, Progress in Inorganic Chemistry, ed. F. A. Cotton, John Wiley and Sons, New York, 1967, vol. 8, p. 391 Search PubMed; (b) N. Dowling, P. M. Henry, N. A. Lewis and H. Taube, Inorg. Chem., 1981, 20, 2345 CrossRef CAS.
- (a) I. Wallace and D. P. Rillema, Inorg. Chem., 1993, 32, 3836 CrossRef CAS; (b) L. Wallace, D. C. Jackman, D. P. Rillema and J. W. Merkert, Inorg. Chem., 1995, 34, 5210 CrossRef CAS; (c) M. M. Richter, J. D. Debad, D. R. Striplin, G. A. Crosby and A. J. Bard, Anal. Chem., 1996, 68, 4370 CrossRef.
- L. O. Ciana and A. Haim, J. Heterocycl. Chem., 1984, 121, 607.
- B. Chaudret, G. Commenges and R. Poilblanc, J. Chem. Soc., Dalton Trans., 1984, 1635 RSC.
- F. A. Cotton and J. G. Norman, J. Coord. Chem., 1971, 1, 161.
- M. Krejik, M. Dank and F. Hartl, J. Electroanal. Chem., 1991, 317, 179 CrossRef CAS.
- R. Hooft, COLLECT, Data Collection Software for Nonius KappaCCD Devices, Nonius B.V., Delft, The Netherlands, 1998 Search PubMed.
- Z. Otwinowski and W. Minor, Processing of X-ray Diffraction Data Collected in Oscillation Mode, eds. C. W. Carter Jr. and R. M. Sweet, Academic Press, New York, 1997, vol. 276, pp. 307–326 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 CrossRef.
- G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2001 Search PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2004 |