Mechanism of the ruthenium-catalyzed formation of pyrane-2-one and their sulfur and selen analogs from acetylene and CX2
(X![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) =O, S, Se): a theoretical study†
=O, S, Se): a theoretical study†
Georg
Dazinger
,
Roland
Schmid
and
Karl
Kirchner
*
Institute of Applied Synthetic Chemistry, Vienna University of Technology, Getreidemarkt 9, A-1060, Vienna, Austria. E-mail: E-mail: kkirch@mail.zserv.tuwien.ac.at
First published on 28th November 2003
Abstract
Complete catalytic cycles for the (actual or potential) cycloaddition of acetylene with CX2
(X![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =O, S, Se), mediated by CpRu(COD)Cl as the precatalyst, are proposed on the basis of DFT/B3LYP calculations. The common initial step is the replacement of labile COD by two molecules of acetylene to afford a bisacetylene complex. Oxidative coupling of the acetylene ligands results in the formation a coordinatively saturated metallacyclopentatriene complex. This metallacycle adds the C
=O, S, Se), mediated by CpRu(COD)Cl as the precatalyst, are proposed on the basis of DFT/B3LYP calculations. The common initial step is the replacement of labile COD by two molecules of acetylene to afford a bisacetylene complex. Oxidative coupling of the acetylene ligands results in the formation a coordinatively saturated metallacyclopentatriene complex. This metallacycle adds the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X bond of CX2 directly to the RuC bond in a concerted fashion, forming an unusual bicyclic carbene intermediate. The activation energies for this addition are 14.2, 15.3 and 30.0 kcal mol−1 for X=O, S and Se, respectively, thus rendering the CSe2 addition the most difficult. In a subsequent reductive elimination a coordinatively unsaturated metallaheteronorbornene intermediate is formed, which eventually rearranges into RuCpCl complexes bearing pyrane-2-one, thiopyrane-2-thione and selenopyrane-2-selenone ligands coordinated in an η4 fashion. The energy barrier for the reductive elimination step decreases in the order O>S>Se, being 28.1, 17.7 and 14.6 kcal mol−1, respectively. Overall, the reaction with CS2 is thus the most favorable. Completion of the cycles is achieved by an exothermic displacement of the respective heterocyclic product by two acetylene molecules, regenerating the bisacetylene complex.
X bond of CX2 directly to the RuC bond in a concerted fashion, forming an unusual bicyclic carbene intermediate. The activation energies for this addition are 14.2, 15.3 and 30.0 kcal mol−1 for X=O, S and Se, respectively, thus rendering the CSe2 addition the most difficult. In a subsequent reductive elimination a coordinatively unsaturated metallaheteronorbornene intermediate is formed, which eventually rearranges into RuCpCl complexes bearing pyrane-2-one, thiopyrane-2-thione and selenopyrane-2-selenone ligands coordinated in an η4 fashion. The energy barrier for the reductive elimination step decreases in the order O>S>Se, being 28.1, 17.7 and 14.6 kcal mol−1, respectively. Overall, the reaction with CS2 is thus the most favorable. Completion of the cycles is achieved by an exothermic displacement of the respective heterocyclic product by two acetylene molecules, regenerating the bisacetylene complex.
Introduction
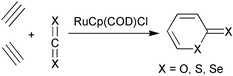
The seminal tool that the chemist uses to systematize elemental relationships is the periodic table. Its function is to serve as a framework, as an organizational outline , for the vast and widely diversified information of chemistry. By analyzing the changes in reactivity down a group the behavior of the individual elements is more fully appreciated. This is all the more relevant since it is commonly a combination of factors that are responsible for a particular outcome, for instance, why HF is a much weaker acid than HCl,1 or why BF3 is a weaker Lewis acid than BCl3.2In the present contribution we focus on the cyclotrimerization of two alkynes with an unsaturated compound, catalyzed by RuCpCl, according to Scheme 1.
 | ||
| Scheme 1 | ||
Of these transformations, the reaction with CS2 has been realized, in fact, recently by Itoh and coworkers, resulting in thiopyrane-2-thiones.3 Likewise, isocyanates4 and isothiocyanates3 have been reacted, giving pyridine-2-ones and thiopyrane-2-imines. Such transition-metal-mediated C–S bond formations would seem highly unusual in view of the strong metal–sulfur bonds prone to deactivate the catalyst. In fact, so far only the ruthenium complexes RuCp(COD)X and RuCp*(COD)X (X=Cl, Br) have proved to be suited for this undertaking.
For the underlying reaction mechanism, Itoh et al.3 invoked a metallacyclopentadiene as a key intermediate. Detailed DFT calculations, however, from our collaboration on similar systems point to the intermediacy of a metallacyclopentatriene complex that is able to easily add a CS bond of CS2 in a concerted [2+2] fashion directly to the RuC double bond.5,6 Based on this experience we will investigate here, by means of DFT/B3LYP calculations,7,8 whether the analogous insertions of CO2 and CSe2 are conceivable and what atomic properties determine the reactivities. A better understanding of such cyclotrimerizations is desirable since these processes are not only metal-economic but also synthetically useful for obtaining six-membered heterocyclic systems.9–12 In addition, carbon dioxide is an abundant source of carbon.
Results and discussion
The cyclotrimerization of two molecules of acetylene and CS2 at RuCp(COD)Cl has been analyzed in detail previously.6 The initial step is the replacement of labile COD in the Ru complex by two molecules of acetylene to afford, via the bisacetylene complex A, the metallacyclopentatriene B as a result of facile oxidative coupling (Scheme 2, energies in kcal mol−1). | ||
| Scheme 2 | ||
In analogy to CS2, B also easily adds one CO bond of the incoming CO2 molecule in an almost concerted [2+2] fashion directly to the RuC bond to afford the bicyclic intermediate C. This is slightly endothermic, requiring an activation energy of 14.2. kcal mol−1
(cf. slightly exothermic with an activation energy of 15.3 kcal for CS2). The energetic profile and the geometries involved are shown in Scheme 3 and Fig. 1. In C, two new bonds, Ru–O (2.09 Å) and C–C (1.57 Å), are formed. This is noteworthy since in the transition state TSBC the carbon dioxide is close to η1-coordinated with the C⋯C distance still being very long (2.10 Å), indicating an, at best, weak interaction. The two CO distances are different because the binding of O to Ru weakens the adjacent CO bond. In going to C, the latter becomes a C–O single bond, while the terminal C–O bond is also weakened since the carbon atom is involved in an additional bond. Both C–O distances remain largely unchanged in the subsequent reactions.
 | ||
| Scheme 3 | ||
 | ||
| Fig. 1 Optimized B3LYP geometries of the equilibrium structures C, D, E and the transition states TSBC, TSCD, TSDE (distances in Å) for the reaction of B with CO2. | ||
In the onward reaction of C the Ru–O bond is eventually broken and concomitantly a C–O bond to the other α carbon of the metallacycle is formed. The transformation of C to D is essentially a reductive elimination in which a vacant coordination site is created and D may be considered as an unsaturated metallaheteronorbornene complex. If the incoming ligand is an alkene6 this vacant site becomes involved in an agostic interaction. In the present case of CO2 attack, of course, no such possibility is available apart from a weak Ru–O interaction with the unfavorably positioned oxygen electron lone pair. Therefore, the reaction of C to D is appreciably endothermic (18.0 kcal mol−1) with a relatively high activation energy (28.1 kcal mol−1). In the case of CS2 a much smaller activation energy of 17.7 kcal mol−1 is needed for the reductive elimination. This difference may be traced to the different atomic radii of O and S (0.66 vs. 1.07 Å13) controlling the interatomic interactions. The transition state TSCD largely resembles D, having an oxygen atom in a pyramidal environment with the C–O bond partly formed and the Ru–O bond already weakened (0.21 Å longer than in C). The C–O bond in D is already a normal bond, although on the long side (1.48 Å). It becomes a typical C–O bond (1.39 Å) in the final species E, when the Ru–O bond has completely broken and ruthenium binds an η4-pyrane-2-one. The transformation of D into E starts with pivoting of the butadiene moiety, moving it from di-σ- to η4-coordinated as in the CS2 system. Completion of the cycle is achieved via the reaction E+2 HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH→pyrane-2-one+A, which is exothermic by 14.7 kcal mol−1
(cf. 17.9 kcal mol−1 for the CS2 system).
CH→pyrane-2-one+A, which is exothermic by 14.7 kcal mol−1
(cf. 17.9 kcal mol−1 for the CS2 system).
Let us now turn to the reaction of B with CSe2. The energetic profile and the geometries of the species involved are displayed in Scheme 4 and Fig. 2, respectively. While the intermediates C, D and E and the transition states connecting them are very similar to the CO2 and CS2 analogs, the energies involved are not. Note that the activation energy required for inserting the CSe bond into the RuC bond of B is twice as high (30.0 kcal mol−1vs. 14.2 for CO2 and 15.3 for CS2). This implies that CSe2 is a much weaker nucleophile towards ruthenium. The catalytic cycle is completed via the reaction E+2 HCCH→selenopyrane-2-selenone+A, which is exothermic by 17.1 kcal mol−1. The catalytic cycle for the addition of CX2
(X=O, S, Se) to B is shown in Scheme 5.
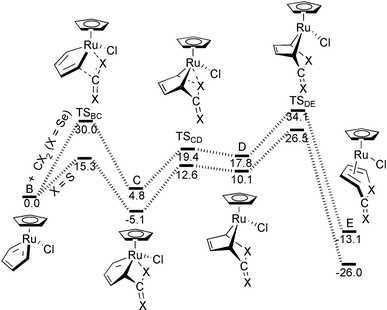 | ||
| Scheme 4 | ||
 | ||
| Fig. 2 Optimized B3LYP geometries of the equilibrium structures C, D, E and transition states TSBC, TSCD, TSDE (distances in Å) for the reaction of B with CSe2. | ||
 | ||
| Scheme 5 | ||
Conclusions
According to the present DFT/B3LYP calculations the mechanisms of the hypothesized cyclotrimerization of alkynes with CO2, CS2 and CSe2 catalyzed by the CpRuCl fragment should be very similar. The metallacyclopentatriene B, appearing as a common key intermediate from oxidative coupling of two alkyne ligands, is capable of adding CX2 in a concerted fashion to the RuC bond to give a new bicyclic carbene intermediate C. However, there are notable energetic differences. Whereas the barrier for the [2+2] addition of a CX bond to the RuC bond of B is low in the case of X=O and S, for X=Se the addition is the rate-limiting step. Conversely, the subsequent reductive elimination (C→D) is facile for X=S and Se but difficult for X=O, in which case the transformation of a Ru–O into a C–O bond is unfavorable and constitutes the rate-determining step. Therefore, the overall reaction with CS2 is the most favorable.
Theoretical
All calculations were performed using the Gaussian 98 software package14 on a Silicon Graphics Cray Origin 2000 at the Vienna University of Technology. The geometry and energy of the model complexes and the transition states were optimized at the B3LYP level with the Stuttgart/Dresden ECP (sdd) basis set15 to describe the electrons of the ruthenium atom. For all other atoms the 6-31G** basis set was employed.16 Frequency calculations were performed to confirm the nature of the stationary points, yielding one imaginary frequency for the transition states and none for the minima. Each transition state was further confirmed by following its vibrational mode downhill on both sides and obtaining the minima presented on the reaction energy profiles. All geometries were optimized without constraints (C1 symmetry) and the energies were zero-point corrected. Relative energies were compared taking into account the total number of molecules present.Acknowledgements
Financial support by the “Fonds zur Förderung der wissenschaftlichen Forschung” is gratefully acknowledged (Project No. P14681-CHE).References
- R. Schmid and A. M. Miah, J. Chem. Educ., 2001, 78, 116 CAS.
- H. Hirao, K. Omoto and H. Fujimoto, J. Phys. Chem. A, 1999, 103, 5807 CrossRef CAS.
- Y. Yamamoto, H. Takagishi and K. Itoh, J. Am. Chem. Soc., 2002, 124, 28 CrossRef CAS.
- Y. Yamamoto, H. Takagishi and K. Itoh, Org Lett., 2002, 13, 2117.
- M. J. Calhorda, K. Kirchner and L. F. Veiros, in Perspectives in Organometallic Chemistry, eds. C. G. Screttas and B. R. Steele, The Royal Society of Chemistry, Cambridge, 2003, p. 111–119 Search PubMed.
- K. Kirchner, M. J. Calhorda, R. Schmid and L. F. Veiros, J. Am. Chem. Soc., 2003, 125, 11721 CrossRef CAS.
- R. G. Parr and Y. Wang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett, 1989, 157, 200 CrossRef CAS; (b) C. Lee, W. Yang and G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- (a) P. Hong and H. Yamazaki, Tetrahedron Lett., 1977, 18, 1333 CrossRef; (b) P. Hong and H. Yamazaki, Synthesis, 1977, 50 CrossRef CAS; (c) R. A. Earl and K. P. C. Vollhardt, J. Org. Chem., 1984, 49, 4786 CrossRef CAS; (d) P. Diversi, G. Ingrosso, A. Lucherini and S. Malquori, J. Mol. Catal., 1987, 40, 267 CrossRef CAS.
- (a) H. Hoberg and B. W. Oster, Synthesis, 1982, 324 CrossRef CAS; (b) H. Hoberg and B. W. Oster, J. Organomet. Chem., 1982, 234, C35 CrossRef CAS; (c) H. Hoberg and B. W. Oster, J. Organomet. Chem., 1983, 235, 359 CrossRef CAS.
- S. T. Flynn, S. E. Hasso-Henerson and A. W. Parkins, J. Mol. Catal., 1985, 32, 101 CrossRef CAS.
- (a) Y. Wakatsuki and H. Yamazaki, J. Chem. Soc., Chem. Commun., 1973, 280 CAS; (b) H. Yamazaki, J. Synth. Org. Chem. Jpn., 1987, 45, 244 Search PubMed.
- C. H. Suresh and N. Koga, J. Phys. Chem. A, 2001, 105, 5940 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle and J. A. Pople, GAUSSIAN 98 (Revision A.7), Gaussian, Inc., Pittsburgh, PA, 1998 Search PubMed.
- (a) U. Haeusermann, M. Dolg, H. Stoll, H. Preuss, P. Schwerdtfeger and R. M. Pitzer, Mol. Phys., 1993, 78, 1211; (b) W. Kuechle, M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1994, 100, 7535 CrossRef; (c) T. Leininger, A. Nicklass, H. Stoll, M. Dolg and P. Schwerdtfeger, J. Chem. Phys., 1996, 105, 1052 CrossRef CAS.
- (a) A. D. McClean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS; (b) R. Krishnan, J. S. Binkley, R. Seeger and J. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS; (c) A. J. H. Wachters, J. Chem. Phys., 1970, 52, 1033 CrossRef CAS; (d) P. J. Hay, J. Chem. Phys., 1977, 66, 4377 CrossRef CAS; (e) K. Raghavachari and G. W. Trucks, J. Chem. Phys., 1989, 91, 2457 CrossRef CAS; (f) M. P. McGrath and L. Radom, J. Chem. Phys., 1991, 94, 511 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: coordinates and total energy of the complexes C, D, E and transition states TSBC, TSCD, TSDE. See http://www.rsc.org/suppdata/nj/b3/b307843d/ |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2004 |