Microwave heating of heterogeneously catalysed Suzuki reactions in a micro reactor
Ping
He
,
Stephen J.
Haswell
* and
Paul D. I.
Fletcher
Department of Chemistry, The University of Hull, Hull, UK HU6 7RX. E-mail: S.J.Haswell@hull.ac.uk
First published on 2nd December 2003
Abstract
The Suzuki cross-coupling reaction of aryl halides with phenylboronic acid to form biaryls has been used to illustrate the development of a microwave based technique capable of delivering heat locally to a heterogeneous Pd-supported catalyst located within a micro reactor device. A 10–15 nm gold film patch, located on the outside surface of the base of a glass micro reactor, was found to efficiently assist in the heating of the catalyst when irradiated with 5–7 W of microwave power at 2.45 GHz. Using a hydrodynamically pumped system, reactant–catalyst contact times of less than 60 s were found to give conversions for different substrates which were in the range 50–99%. Two methods of loading catalysts into the micro reactor were investigated which required either 1.5 or 6 mg of material.
Introduction
To date the majority of liquid based chemical syntheses carried out in lab-on-a-chip micro reactors have involved homogeneous reactions performed at room temperature.1 As discussed in ref. 1, the ability to spatially control localized concentrations of reactant, intermediates and products within the micro-channel networks of micro reactors enables a level of reaction control (including rates, yields and product selectivities) which is not achievable in bulk reactors where concentrations are generally uniform. In addition to the control of local concentrations, the ability to deliver localized heating2–3 and the more effective use of in situ supported reagents such as catalysts would add a further dimension of reaction control and hence extend the range of potential applications of micro reactors. Accordingly, in this paper we describe a microwave (MW) based localized heating technique used in conjunction with an immobilized palladium catalyst to perform a number of exemplary Suzuki based reactions4 within a hydrodynamically pumped micro reactor.The palladium-catalyzed cross coupling of aryl halides with aryl boronic acids was selected for this study because of its relevance as a popular method for the selective formation of carbon–carbon bonds4–5 (see Scheme 1). Such Suzuki reactions are commonly performed using a homogeneous soluble palladium catalyst, however, recovery of such homogeneous catalysts can prove difficult at the aqueous work-up stage.6 Developing a methodology based on a heterogeneous catalyst in a continuous flow micro reactor type operation offers many advantages over homogeneous batch based methods. These include effective product isolation from the catalysts reaction zone and improved control of the catalyst–reactant contact time leading to more rapid optimization of both yield and product selectivity. In addition, the solid phase catalysts which are required only in mg quantities, can be readily tested under reaction conditions and are easily recovered for reuse if required.
 | ||
| Scheme 1 General reaction scheme for palladium catalysed Suzuki cross-coupling. | ||
The use of MW heating in organic synthesis7–8 including Suzuki reactions9–11 has attracted considerable interest in the past few years where the main benefits are reported to be significant rate-enhancements and increased product yields.11–13 These features have attracted interest from the drug discovery and medicinal chemistry communities14–15 who are also interested in the high throughput, rapid optimization, small volume and intrinsically safe operating characteristics of micro reactors. Coupling the use of MW heating with micro reactor based synthesis potentially combines a number of advantages of each technology and hence may have good potential in applications involving the synthesis of fine chemicals. One obvious problem with the direct heating of liquid phase reagent solution in a micro reactor channel of micron depth is the limited absorption of MW energy directly by the channel contents. Even for polar species which show (relatively) strong absorption of MW, efficient absorption typically requires a liquid depth in the order of a cm for MWs at 2.45 GHz.16 However, this shortcoming can be overcome by exploiting the absorption of MWs by either supported metal catalysts within a channel and/or thin metallic films16 located on the surface of a micro reactor. Positioning of the MW absorbing species or patch in or on the micro reactor enables localized heating over a prescribed length of a channel and, through careful control of the substrate amount/thickness, selected local temperatures may be reliably generated.16 In the present study, direct MW heating of a catalyst and the combined MW heating of a catalyst and a thin gold film on the outer surface of the micro reactor are evaluated as methodologies for the efficient localized heating of a micro reactor device.
Experimental
The reactions were conducted in glass micro reactors supplied by Micro Chemical Systems Ltd (Hull). Two linear channel designs (see Fig. 1) were used in which a premixed reactant solution containing an aryl halide (0.1 M), phenylboronic acid (0.12 M) and K2CO3 (0.3 M) with dimethylformamide (DMF) and H2O (3:1 volume ratio) mixture as solvent, was pumped from an input reservoir to a collection reservoir through a catalyst bed. PEEK tubing (outer diameter OD = 1.58 mm, inner diameter ID = 0.18 mm) was fitted to the inlet and outlet reservoirs and sealed using Torr seal (Varian). The end of the inlet tube was connected to a Harvard PHD 2000 syringe pump which delivered the homogeneous reaction solution to the reactor. The end of the outlet tube was connected to a sample vial which was placed in an ice bath and used to collect the reaction products.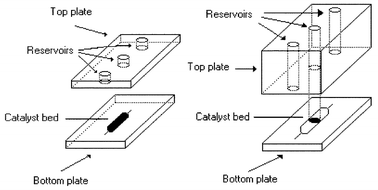 | ||
| Fig. 1 Schematic diagram of the linear channel micro reactor designs and catalyst packing strategies used to perform Suzuki based reactions. | ||
In design A, the top and bottom plates of the micro reactor were both 3 mm thick. The catalyst channel was 1.5 mm wide, 80 µm deep and 15 mm long and connected to the inlet and outlet reservoirs by channels which were 130 µm wide and 50 µm deep. Design B used a 10 mm thick top plate and a 3 mm bottom plate with a catalyst channel which was 1.5 mm wide, 50 µm deep and 15 mm long, i.e. 20 µm shallower than that of design A. The catalyst channel again used channels which were 130 µm wide and 50 µm deep to connect to the inlet and outlet reservoirs. For both micro reactor designs, the catalyst was introduced as a dry powder through the central port (3 mm in diameter) and then located in the micro reactor channel using the air pressure generated from a hand held syringe. This packing procedure only took a few minutes to complete, as did the removal of the wet catalyst achieved by drawing the slurry out through the central port again with a syringe. During reaction, the central port was blanked off with a tightly fitting PEEK rod. The catalyst consisted of particles of size 45–63 µm (selected by sieving). These catalyst particles fitted within the catalyst channel of design A to create a monolayer of particles (requiring 1.5 mg) over the entire catalyst channel area. During the continuous flow maintained during reaction, the particles were retained by the keystone effect.17 The catalyst particles were too large to fit within the depth of the catalyst channel of design B and were located within the central reservoir in the form of a plug about 0.5–1 mm in height (requiring 6 mg). As required, an area (15 × 1.5 mm) on the outside of the bottom plate corresponding to the region of catalyst packing was sputter coated with gold to a thickness of either 10 or 15 nm using a SEMPREP 2 Sputter Coater (Nanotech Ltd.). During MW irradiation no electrical sparking was observed from the film, however, caution may be required if thicker or larger areas of metalization are used.
The micro reactor was heated in the cavity of a Discover MW system from CEM, which is capable of delivering 0–300 W of MW power at 2.45 GHz. An IR temperature sensor located in the base of the Discover enabled determination of the temperature at the base of the micro reactor. It should be emphasized that this temperature refers to the lower exterior surface of the micro reactor and not the actual reaction zone within the catalyst channel. The measured temperature of each reaction was mediated by variation of the MW heating power (power time mode) in conjunction with different solution flow rates. The residence times of the solutions within the catalyst bed were measured by timing the movement of the liquid front during first filling.
During a reaction run, product samples were collected in a cooled (0 °C) product vial for 5 min. This collection period was found to be long enough to obtain a representative sample for subsequent analysis. Samples were weighed, a known amount of dodecane was added as an internal standard and then treated with 1 M aqueous NaOH to remove unreacted phenylboronic acid. The remaining organic material was then extracted, washed three times with distilled water, collected and dried over MgSO4. Samples were then analysed for both aryl halide reactant and biaryl product using a GC instrument (Shimadzu GC-17A) equipped with a capillary column (CP SIL 8 CB, 30 m length, Chrompack). Pressure of carrier gas (helium) was 600 kPa and injector temperature was set to 250 °C. The GC column temperature was held initially at 70 °C for 4 min, ramped at 20 °C min−1 to reach 240 °C which was then held for 12 min. The retention times for starting materials and products are summarized in Table 1.
| Compound | Retention time/min |
|---|---|
| 4-Bromobenzonitrile | 9.4 |
| 4-Cyanobiphenyl | 13.2 |
| 4-Bromonitrobenzene | 10.3 |
| 4-Nitrobiphenyl | 14.0 |
| 4-Bromobenzaldehyde | 9.2 |
| 4-Biphenylaldehyde | 13.1 |
| 4-Iodoanisole | 10.0 |
| 4-Methoxybiphenyl | 12.6 |
| 4-Iodotoluene | 8.4 |
| 4-Methylbiphenyl | 11.5 |
| Benzonitrile | 6.5 |
| Biphenyl | 10.6 |
| Dodecane | 8.8 |
The retention times for all compounds were verified using pure materials obtained as indicated below. All analysis was carried out in duplicate with replicates agreeing to within a 5% variation. Conversions were calculated from GC data by determining the quantity of aryl halide present in the collection vial after reaction using internal standard method. Overall mass balances were checked using the GC data for the product and were correct to within a few percent.
The reagents and solvents 4-bromobenzonitrile (Aldrich, 99%), 4-cyanobiphenyl (Aldrich, 99%), 4-bromonitrobenzene (Aldrich, 99%), 4-nitrobiphenyl (Aldrich, 99%), 4-bromobenzaldehyde (Aldrich, 99%), biphenylaldehyde (Aldrich, 99%), 4-iodoanisole (Aldrich, 98%), 4-methoxybiphenyl (Aldrich, 99%), 4-iodotoluene (Aldrich, 99%), 4-methylbiphenyl (Aldrich, 99%), benzonitrile (Aldrich, 99%), biphenyl (Aldrich, 99%), phenylboronic acid (Aldrich, 97%), dodecane (Aldrich, 99%), N,N-dimethylformamide (DMF, Fluka, 99%), dichloromethane (DCM, Fisher, 99%), MgSO4 (Fisher, 99%) and K2CO3 (Lancaster, 99%) were used without further purification. The catalysts Pd supported on alumina (Pd/Al2O3) and Pd anchored on polymer (Pd–polymer) were supplied by Johnson Matthey and contained 5 wt% Pd for the Pd/Al2O3 and 4 wt% Pd for Pd–polymer.
Results and discussion
The first set of experiments were carried out using the micro reactor with a catalyst plug (design B) and 5 selected substrates, in order to evaluate the potential of the proposed methodology. It should be noted that, in order to maintain comparable temperature ranges for each reaction, monitored by the optical detector in the base of the Discover instrument, the applied MW power was varied. As shown in Fig. 2, the gold films became less efficient at absorbing the MW power and reaching a high temperature following MW exposure during a larger number of 5 min heating cycles. | ||
| Fig. 2 Variation of the MW absorption efficiency (expressed as the ratio IR sensor temperature/MW power) following repeated 5 min heating cycles. The gold film was 15 nm thick initially. | ||
The appearance of the gold film changed from dark grey to an increasingly lighter grey following repeated heated cycles. This observation suggested that the films became progressively thinner, probably due to evaporation of the gold, rather than “peeling” away from the glass surface of the micro reactor. For the experiments here, the first heating cycle was ignored and all reaction measurements refer to heating cycles 2–4 where the gold films are more constant. As noted earlier, the MW power was adjusted for each run so as to obtain a constant temperature as measured by the IR sensor.
Some preliminary measurements were made using heating patch films of different materials. For the same MW power and film thickness, the measured temperatures were in the order Au > Pt ≈ Pd > C. This ranking sequence correlates with the electrical conductivities of the materials, i.e. the more conducting, the better the MW absorption and the higher the temperature reached. It was observed that improved MW heating could be obtained with Pt by using thicker films. Further work is proceeding to find the best choice of metal material and film thickness which optimizes heating performance, metal film stability and its adhesion to the glass surface of the micro reactor. The results presented in Table 2 for different aryl halide reactants indicate that deactivated species such as 4-bromonitrobenzene and 4-bromobenzonitrile are more reactive than activated substrates and give correspondingly high conversions. This is consistent with a mechanism in which electron withdrawing groups favor oxidative addition of aryl halides to a Pd(0) species.2
| Aryl halide | ||||
|---|---|---|---|---|
| X | R | MW power/W | Temp/°C | Conva% |
| a Conversions were calculated from GC data based on the amount of aryl halide present in the collection vial after reaction. Only cross-coupling product was produced. | ||||
| Br | NO2 | 50 | 90–98 | 98 |
| Br | CN | 55 | 90–102 | 99 |
| Br | CHO | 40 | 90–96 | 75 |
| I | OCH3 | 80 | 90–100 | 75 |
| I | CH3 | 55 | 90–98 | 58 |
Having established that the proposed methodology was able to generate significant product, the effects of different heating methods on product yield were measured using 4-bromobenzonitrile as aryl halide reactant. As seen in Table 3, no conversion is detected under the reaction conditions used at room temperature. In the second heating method, the micro reactor was immersed in an oil bath at 130 °C. Reactants from a reservoir at room temperature were then pumped through the micro reactor and a 65% conversion was obtained. The third heating method used the MW absorption by the alumina supported catalyst without any metal film heating patch. In this case, 150 W of MW power were required to produce a temperature of around 95 °C as sensed on the external micro reactor surface by the IR sensor. The localized temperature at the catalyst surface is hard to estimate under these conditions as it is simultaneously cooled by the reagent flow and heated by MW absorption into (mainly) the alumina catalyst support. In the fourth method, MW energy was absorbed into both the catalyst and the gold film heating patch situated underneath the catalyst channel. The more efficient MW absorption is clearly evident by the reduced MW power required to achieve the measured temperature of around 95 °C. In all cases a flow rate of 5 µl min−1 was used and it can be seen that the best conversion (virtually 100%) is obtained at relatively low MW power when MW absorption into both the catalyst and gold coating is used. Because of the coupled effects of reagent flows, energy absorption and conduction, it is difficult to estimate the localized temperature within the reaction zone. On the basis that the conversion is an effective measure of the localized temperature experienced by the reaction (i.e. the MW effect is purely thermal), the data of Table 3 suggest that MW absorption into both catalyst support and gold film is both effective and efficient in focusing heating energy into the small reaction zone within the micro reactor.
| Heating method | MW power/W | Temp/°C | Conva % |
|---|---|---|---|
| a Conversions were calculated from GC data based on the amount of 4-bromobenzeonitrile present in the collection vial after reaction. Only cross-coupling product was produced. | |||
| Room temperature | 0 | 25 | 0 |
| Oil bath | 0 | 130 | 65 |
| MW heating only | 150 | 94–98 | 71 |
| MW heating plus gold coating | 55 | 90–102 | 99 |
A comparison between the two catalyst packing modes (design A and B) was carried out in order to evaluate the proposed methodology. As seen in Table 4, product yields were generally similar for the two packing designs. However design A, using a slightly longer residence time (lower flow rate), did need less mass of catalyst and was found to require significantly less MW energy. Although somewhat uncertain due to the variation in gold film properties (Fig. 2), the apparently more efficient heating of design A is attributed to the fact that the catalyst is present as a thin particle monolayer to which heat transfer from the gold patch is more effectively achieved. The results also indicate that a system using a catalyst particle monolayer design plus a relatively low powered MW system (5 W, see Table 4) can be used to achieve good product yields in systems requiring localized temperatures in excess of 100 °C. Noting that the present system is very far from being optimized, these first results are very encouraging.
| Substrate | Flow/µl min−1 | MW power/W | Packing design | Conv % |
|---|---|---|---|---|
| /Contact time/s | /Temp/°C | |||
| a A 10 nm gold film was used for this run which is therefore not strictly comparable. Use of a 15 nm gold film at a measured temperature range of 90–103 °C achieved with 10 W MW power gave an overall conversion of 98%. However, 50% of the conversion was biphenyl side product. | ||||
| Br–C6H4–NO2 | 5/36 | 50/90–98 | B | 98 |
| Br–C6H4–NO2 | 3/44 | 7/90–105 | A | 90 |
| Br–C6H4–CN | 5/36 | 55/90–102 | B | 99 |
| Br–C6H4–CN | 3/44 | 5/90–108 | A | 92 |
| Br–C6H4–CHO | 5/36 | 40/90–96 | B | 75 |
| Br–C6H4–CHO | 3/44 | 50/80–90 | A | 72a |
Finally, in order to evaluate the suitability of the proposed packing methodology to deal with polymeric based catalysts, a Pd anchored polystyrene support was used in a B design micro reactor. Using the design B micro reactor with the substrate 4-bromobenzonitrile, reagent concentrations as listed in Tables 2–4, 3 mg of both catalysts, 3 µl min−1 flow rate, a catalyst contact time of 32 s and a MW power of 55 W giving a measured temperature range of 90–100 °C which gave a 75% conversion for the Pd–polystyrene and 72% for Pd–alumina. Despite having a 20% lower loading of metal than the alumina supported catalyst, the Pd–polystyrene catalyst gives a comparable product yield. When using the polymer supported catalyst, it was found that filling the channel of micro reactor design A was practically very difficult due to the particle shape, size and texture however, filling design B with the polymeric based catalyst was virtually straightforward.
Conclusions
In order to more fully exploit the use of micro reactor technology in chemical and biochemical synthesis, methods for the controlled localized heating and the utilization of highly intensive surface properties need to be established. Whilst micro reactor based heating may be achieved using for example localized electrical resistance,2–3 in this work a contactless technique based upon MW induced heating has been used. In this way the controlled heating of a reaction can be achieved using selective MW absorption into both a catalyst and an externally applied gold film. It was found that at present the 10–15 nm film of gold used, whilst mechanically stable, did appear to evaporate and work in this area to generate a more robust film is in progress. In addition, a simple and practical technique is described for the loading and removal of an alumina and polymer supported metal catalyst from a micro reactor device. Whilst a Suzuki type reaction was used to illustrate the methodology, the results clearly point the way to developing a wider range of chemical methods and process applications of micro reactor technology.Acknowledgements
We thank CEM Microwave Technology Ltd. and the Engineering and Physical Sciences Research Council for funding and Mr. Sinclair of the University of Hull for the preparation of the gold coatings.References
- P. D. I. Fletcher, S. J. Haswell, E. Pombo-Villar, B. H. Warrington, P. Watts, S. Y. F. Wong and X. Zhang, Tetrahedron, 2002, 58, 4735 CrossRef CAS.
- M. U. Kopp, A. J. de Mello and A. Menz, Science, 1998, 1046 CrossRef CAS.
- D. S. Yoon, Y-S. Lee, Y. Lee, H. J. Cho, S. W. Sung, K. W. Oh, J. Cha and G. Lim, J. Micromech. Microeng., 2002, 12, 813 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
- S. Kotha, K. Lahiri and D. Kashinath, Tetrahedron, 2002, 58, 9633 CrossRef CAS.
- J. H. Clark, Green Chem., 1999, 1, 1 RSC.
- P. Lidstrom, J. Tierney, B. Wathey and J. Westman, Tetrahedron, 2001, 57, 9225 CrossRef CAS.
- L. Perreux and A. Loupy, Tetrahedron, 2001, 57, 9199 CrossRef CAS.
- C. G. Blettner, W. A. Konig, W. Stenzel and T. Schotten, J. Org. Chem., 1999, 64, 3885 CrossRef CAS.
- N. E. Leadbeater and M. Marco, J. Org. Chem., 2003, 68, 888 CrossRef CAS.
- M. Larhed and A. Hallberg, J. Org. Chem., 1996, 61, 9582 CrossRef CAS.
- D. Villemin and F. Caillot, Tetrahedron Lett., 2001, 42, 639 CrossRef CAS.
- N. Kuhnert and T. N. Danks, Green Chem., 2001, 3, 98 Search PubMed.
- (a) B. Wathey, J. Tierney, P. Lidstrom and J. Westman, Drug Discov. Today, 2002, 7, 373 CrossRef CAS; (b) M. Larhed and A. Hallberg, Drug Discov. Today, 2001, 6, 406 CrossRef CAS.
- J. L. Krstenansky and I. Cotterill, Curr. Opin. Drug Discov. Dev., 2000, 4, 454 Search PubMed.
- P. He, P. D. I. Fletcher and S. J. Haswell, Proceedings of Conference and Workshop on Micro Total Analytical and Chemical Systems, April 7–9, 2003, Hull, UK.
- G. A. Lord, D. B. Gordon, P. Myers and B. W. King, J. Chromatogr., A, 1997, 768, 9 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2004 |