Polymer microfluidic chip for online monitoring of microarray hybridizations
Mikkel
Noerholm
*ab,
Henrik
Bruus
b,
Mogens H.
Jakobsen
a,
Pieter
Telleman
b and
Niels B.
Ramsing
a
aExiqon A/S, Bygstubben 9, DK-2950 Vedbæk, Denmark. E-mail: noerholm@exiqon.com
bMIC – Department of Micro and Nanotechnology, Technical University of Denmark (DTU), DK-2800 Kgs, Lyngby, Denmark
First published on 17th December 2003
Abstract
A disposable single use polymer microfluidics chip has been developed and manufactured by micro injection molding. The chip has the same outer dimensions as a standard microscope slide (25 × 76 × 1.1 mm) and is designed to be compatible with existing microscope slide handling equipment like microarray scanners. The chip contains an inlet, a 10 µL hybridization chamber capable of holding a 1000 spot array, a waste chamber and a vent to allow air to escape when sample is injected. The hybridization chamber ensures highly homogeneous hybridization conditions across the microarray. We describe the use of this chip in a flexible setup with fluorescence based detection, temperature control and liquid handling by computer controlled syringe pumps. The chip and the setup presented in this article provide a powerful tool for highly parallel studies of kinetics and thermodynamics of duplex formation in DNA microarrays. The experimental setup presented in this article enables the on-chip microarray to be hybridized and monitored at several different stringency conditions during a single assay. The performance of the chip and the setup is demonstrated by on-line measurements of a hybridization of a DNA target solution to a microarray. A presented numerical model indicates that the hybridization process in microfluidic hybridization assays is diffusion limited, due to the low values of the diffusion coefficients D of the DNA and RNA molecules involved.
Introduction
Microtechnology found its first application in electronics and in a matter of a few decades revolutionized our daily lives. The concept of miniaturization and functional integration, i.e. the microfabrication of different electronic components and the integration of these components to form complex integrated circuits, is a strategy that can also be used in other fields, e.g. mechanics, optics, chemistry and the life sciences. In 1979, Terry et al. presented “A gas chromatographic air analyser fabricated on silicon wafer using integrated circuit technology”.1 This was the first publication discussing the use of techniques borrowed from microelectronics to fabricate a structure for chemical analysis. The introduction of the concept of micro total analysis systems (μTAS) by Manz and co-workers in 19902 triggered rapidly growing interest in the development of microsystems where all the stages of chemical analysis like sample preparation, chemical reactions, analyte separation, analyte purification, analyte detection, and data analysis are performed in an integrated and automated fashion. The realization of such chemical analysis systems requires miniaturization and integration of a wide variety of components, e.g. mechanic, fluidic, optic, and electronic.Microfabrication (i.e. the fabrication of structures down to micrometers in size) is essential to the development of μTAS. Silicon presented an obvious choice as material for the microelectronics industry due to its semiconductor properties. The explosive growth of microelectronics has led to a wide range of microfabrication tools for silicon and very high levels of experience and expertise exist for working with this material for microtechnology. Silicon is ideal for microfabrication of electronic, mechanic, and optic components and thereby allows for high levels of functional integration. However, the superiority of silicon as a material for μTAS is debatable because the chemical stability of silicon is not very good. In fact many of the microfabrication methods available today are based on the controlled removal of silicon by chemical treatments. Although the surface of silicon can be treated to withstand harsh chemical environments other materials may be more suitable for certain applications. Another important argument for investigating alternative materials is the high cost of silicon, especially in single-use applications, e.g. where μTAS are in contact with biohazardous materials like blood and must be discarded after single use. For these reasons polymers offer interesting alternatives to the use of silicon for μTAS. Compared to silicon, polymers possess a number of attractive qualities for use in chemical or biochemical microsystems, like optical transparency and chemical resistance to aggressive media. Most importantly polymers can easily be machined on the micrometer scale using a number of different methods like milling, laser ablation, hot embossing and injection molding. Another very persuasive argument towards choosing polymers compared to other materials, is the potential for a very low per-unit manufacturing cost, which is attainable when production is scaled up to batch sizes in the range of hundreds of thousands. As the use of polymers for micromechanic, micro-optic, and micro-electronic components is still very much under development, fabrication in this material carries with it concessions to the level of functional integration that can be achieved. Hybrid solutions where microstructures with different functions, fabricated in different materials are assembled to make up a complete μTAS will most likely arise.
In parallel to the rise of interest in μTAS, the concept of DNA microarray technology was conceived by Southern et al. in the early 1990s.3 The hybridization of sample DNA to an array of capture DNA fragments or oligonucleotides immobilized on a solid surface allowed for massive parallel screening. The last couple of years microarray technology has become an important tool in genomics research, satisfying the need for highly parallel analyses needed to exploit the increasing amount of genomic sequence data available. Commercial products have become available to the research community4,5,6 as well as efficient and reliable protocols for printing, hybridization, and scanning of custom-made microarrays based on functionalized glass or polymer slides.7
In this article we present a closed polymer microfluidics chip with temperature control containing a DNA microarray. By combining the fields of microfluidics and microarrays the advantages of both fields can be exploited simultaneously. Microfluidics allows for automated delivery of controlled volumes of sample and reagents to the DNA microarray. Combining fluidic control with accurate temperature control and employing an automated monitoring system for detection of the fluorescent signals from the microarray allows for on-line monitoring of the hybridization of target DNA to capture DNA. Hybridization assays can be repeated automatically choosing different assay parameters like temperature, stringency of the wash buffer, and volume of the wash buffer. Ultimately, this combined microfluidics-microarray system allows for automated selection of optimal assay parameters. This microfluidics-microarray system thereby offers significant improvement over conventional DNA microarray protocols, where a hybridization assay is performed at one particular hybridization temperature and with a fixed set of parameters for post-hybridization washing. Furthermore, the microfluidics-microarray system is able to monitor the kinetics of the DNA hybridization of the chip. Earlier work reporting on the use of microarrays in micro channels has been based on microstructures fabricated in a layer of adhesive tape,8 which does not meet the requirements for quality and reproducibility needed in genomics analyses. Other systems of higher quality have been fabricated by hot embossing, but addressed only a limited number of spots and thus renouncing the highly parallel approach9,10 or they have been using a non-standard detection system.9,11
The chip used in the present work is fabricated by micro injection molding, which yields parts of very high quality and tight tolerances. The integrated hybridization chamber is capable of holding a microarray of 1000 spots, thus insuring highly parallel analysis of a given sample. The on-chip integration of hybridization and waste chamber eliminates the need for cover-slips and open buffer troughs during liquid handling steps, thus assisting in automation and easy handling.
We have used the chip in a flexible setup consisting of a fluorescence microscope, temperature control and liquid handling by computer controlled syringe pumps. We have investigated hybridization of 30-mer targets to an oligonucleotide array of 12- to 20-mer capture probes by pumping various target solutions and wash buffers through the hybridization chamber under different conditions. By controlling the relative pump rate of the two pumps the conditions inside the hybridization chamber can be changed gradually (or incrementally) e.g. by varying the target concentration or the buffer stringency, while at the same time continuously recording the hybridization signal from the array. When the hybridization chamber is filled with a solution of fluorescently labelled target the background signal is, of course, significant. However, as hybridization occurs the increasing concentration of target, at the capture probes on the chamber surface (i.e. in the focal plane of the microscope), is sufficient for the hybridization to be monitored on-line.
Experimental
Materials
All buffers were prepared or diluted using highly purified water (Milli-Q, 18.2 MΩ cm resistivity). 20 × SSC (Saline Sodium Citrate) containing 3 M NaCl and 0.3 M sodium citrate, pH = 7.0 was purchased from Eppendorf and diluted to the appropriate working concentration. To some buffers Tween®20 was added at 0.1% (v/v) indicated by a capital T (e.g. SSCT or Milli-Q/T). Tween®20 was purchased from Riedel-de Haën. To reduce the risk of bubble formation during hybridization, the buffers were freshly degassed by stirring under vacuum (approximately 50 mmHg) at room temperature for a minimum of 30 min.Oligonucleotide design and synthesis
A total of 54 different oligonucleotide capture probes were synthesized using standard phosphoramidite chemistry,12 some containing Locked Nucleic Acid (LNA) substitutions.13 The capture probes were all synthesized with an anthraquinone molecule (AQ) in the 5′-end as described by Koch et al.14 for UV photo immobilization to the polymer chip surface. The capture probes were synthesized with linkers of various designs, e.g. consisting of a stretch of 15 DNA-t monomers (t15) between the AQ and the capture probe sequence. Fluorescence calibration probes were synthesized consisting of a Cy3-fluorophore in the 3′-end and an AQ in the 5′-end connected by a t15-linker. The hybridization data presented in this article are all from a single DNA capture probe with the sequence 5′-AQ-t15-CGCATGGCTTCCATTGGGT-3′ hybridized to a single fluorescently labelled target with the sequence 5′-Cy3-TGCTGGCACCCAATGGAAGCCATGCGCCGG-3′. The results from closer inspection of the data from LNA containing capture probes and different linker designs will be presented elsewhere.Microarray printing
An array of four replicates of 128 spots (i.e. 512 spots) was spotted into the hybridization chamber of the chip with a spot-to-spot distance of 250 µm, using a BioChip Arrayer I (Packard BioChip Technologies, Meriden, CT). The array included spots of various concentrations (0.5–40 µM) of the capture probes, all in 100 mM phosphate buffer (pH = 7.0). After printing of the array the oligos were covalently coupled to the polymer surface by irradiation with 2300 µJ of UV-light (λ ≈ 350 nm) using a Stratalinker 2400 (Stratagene, La Jolla, CA), as described by Koch et al.14 The chips were washed in Milli-Q water for 10 min to remove salt and excess capture probe and spun dry in a centrifuge (5804R, Eppendorf, Hamburg, Germany) at 1000 rpm for 5 min. Finally the chips were closed by lamination with a foil as described in more detail in a later section.Instrumental setup
Two syringe stepper pumps model NE-1000 (New Era Pump Systems, NY, USA) equipped with 1, 2, 5 or 10 mL standard single use syringes were used to introduce liquid into the chip. The two pumps were controlled via a PC running a custom build LabView 6i program (National Instruments Corp., TX, USA) capable of changing the flow rate for each of the syringes individually (0.1–100 µL min−1) according to a table of time point entries, similar to a HPLC gradient system. From each of the two pumps a piece of PTFE-tubing ID 250 µm connected to a 3-way 4-port valve and from here the liquid continued via a piece of tubing into the chip. The tubing length was ∼20 cm and the total dead volume between the valve and the chip was ∼15 µL. Tubing, connectors and valves were purchased from Mikrolab, Aarhus, Denmark. A schematic overview of the experimental setup is shown in Fig. 1A. | ||
| Fig. 1 (A) Schematic overview of the experimental setup. (B) An example of an image that is stitched together from 13 frames acquired by the CCD-camera. The individual frames are faintly visible as vertical lines. The image is placed in an outline of the hybridization chamber to illustrate the position and size of the array in the chamber. The box and vertical line indicate the position of the 50 averaged lines used for the line profiles described in section “Flow experiments”.(C) This cross-section of the chip illustrates how the chip is placed with the foil facing the Peltier element, and the microscope focusing through the chip base on the upper wall of the hybridization chamber where the microarray is printed. In one experiment a temperature probe was introduced through a hole drilled in the chip base to measure the temperature inside the hybridization chamber. Liquid is introduced by syringe pumps via tubing connected to the conical inlet. | ||
The temperature of the hybridization chamber on the chip was controlled by a PE120 Peltier heating and freezing stage (Linkam Scientific Instruments, Surrey, UK) connected to a PC running the control software LinkSys Ver. 2.28. Using this stage and software it was possible to control the temperature according to a table of time point entries with a heating/cooling rate of 0.1 to 10 °C min−1. A reservoir of 5 L cooling water was recirculated through the Peltier stage. In one experiment a TP-100 temperature sensor (Unisense A/S, Aarhus, Denmark) with a probe diameter <100 µm and a T301 thermometer was used to establish the correlation between the temperature inside the hybridization chamber and the temperature set on the Peltier stage. The chip was placed on the Peltier stage which was mounted on a Zeiss fluorescence microscope (Carl Zeiss, Oberkochen, Germany) equipped with an XBO75 lamp, an epifluorescence filter block (FS#20, Zeiss), an objective (5 × /0.25, Fluar, Zeiss), an OptiScan ES102 motorized (x,y)-stage (Prior Scientific Inc., MA, USA), a CoolSNAP CCD camera (Roper Scientific, AZ, USA) and a Uniblitz Shutter (Vincent Associates, NY, USA). The CCD camera, the shutter and the (x,y)-stage were all controlled from a PC running the software MetaVue version 4.6r10 (Universal Imaging Corp., PA, USA). Images were acquired by a journal (a series of commands similar to a macro) created in MetaVue, which acquires a number of frames of the hybridization chamber and moves the (x,y)-stage by exactly the width of a frame between each exposure. The complete hybridization chamber (2.5 × 23 mm) was covered by 13 frames when using the 5× objective. Individual frames were stitched together by the software to provide one large image, which was then saved to the disk. An example of such a stitched image is shown in Fig. 1B. Before stitching, each individual frame was corrected to compensate for differences in illumination across the field of view. This was done by normalizing each frame to a pre-recorded frame acquired of a uniformly fluorescent sample. Repeatability between experiments was ensured by storing the acquisition settings in a file (exposure time, binning, shading correction), which was loaded by the journal at the beginning of each experiment. The journal was run repeatedly at different times (typically every 1–5 min) to obtain a time-lapse series of stitched images that reflects the hybridization events taking place on the chip. By nesting journals within journals it was possible to vary the intervals at which images were acquired in order to collect more data during interesting periods of the experiment and to save disk space during other periods. Typically an experiment generated several hundreds of stitched images that each were a couple of megabytes in size.
Data analysis
The stitched images were analysed using the batch processing feature of the standard microarray analysis software ArrayVision 6.0 Rev.3 (Imaging Research Inc, ON, Canada) as described in the manual for this software. Spot intensity values were recorded using the volume measurement setting in the software rather than density because this method is less sensitive to variations in spot size and position. In all experiments the background was measured locally as the intensity around individual spots and subtracted from the spot signals.Flow experiments
An experiment was performed where a clear solution (Milli-Q) and a BPB-dyed solution (Bromo Phenol Blue in 0.1 × SSC, pH = 7.0) were delivered from the two pumps, at a flow rate of 10 µL min−1. The chip was placed on a light table and pictures were acquired of the hybridization chamber, with the different mixtures of dye and clear solution flowing through, using a standard CCD camera. Images were acquired at the end of each of the conditions in the following series: (A) 100% Milli-Q water for 3 min, (B) 100% BPB-solution for 3 min, (C) 100% Milli-Q water for 3 min, (D) 100% Milli-Q water for additional 2 min and (E) 50% BPB-solution for 5 min. The images were imported into the software SPIP Version 2.32 (Scanning Probe Image Processor, Image Metrology, Lyngby, Denmark) and line profiles were acquired in this program using an average of 50 lines perpendicular to the hybridization chamber (cf.Fig. 1B).Hybridization experiments
The chip was placed in the microscope with the foil facing the Peltier element (set to 30 °C) and with a tube connecting the hybridization chamber to the syringe pumps as illustrated in Fig. 1C. One of the syringe pumps (#1) were equipped with a standard 2 mL disposable syringe containing a 0.01 µM solution of the target in 5× SSCT. The other pump (#2) was equipped with a 2 mL disposable syringe containing pure 5× SSCT. Hybridizations were carried out by first flushing the hybridization chamber with 5× SSCT from pump #2 at 5 µL min−1 and then switching to 5 µL min−1 target solution from pump #1 at experiment start. After 300 min (5 h) of hybridization the flow was switched back to 5× SSCT from pump #2 for washing. In experiments where the hybridization chamber is continuously flushed with target or wash solution for 12–14 h the total volume is in the milliliter range, which cannot be contained in the waste chamber. The excess waste is therefore allowed to exit through the vent in the distal end of the waste chamber. Images were acquired during the entire experiment at intervals of 5–30 min.Results and discussion
Chip design and fabrication
The chip developed in the current work was designed to be compatible with existing microscope slide handling equipment, i.e. it has the same outer dimensions as a standard 1 × 3 inch slide (25 × 76 mm) with a total thickness of 1.1 mm. The chip consists of a 1 mm thick base, containing the microfluidic structures and the hybridization chamber, into which a microarray of up to of ∼1000 spots can be printed. Subsequent to spotting the microarray the chip base is sealed with a 100 µm thick foil covering the whole base. The closed hybridization chamber is connected to the outside via a microfluidic channel to an inlet port.In the present work we have connected the inlet port to two syringe pumps capable of delivering sample and wash buffers to the hybridization chamber. The chip is made of a transparent optical grade polymer to facilitate fluorescence detection of the reactions inside the hybridization chamber. We have used a fluorescence microscope to measure fluorescence intensities because the liquid connections from the pumps to the chip require a flexible setup with room for the necessary tubing. In other applications where target solutions are loaded manually it is perfectly feasible to use a standard microarray scanner, which is available in many modern laboratories working with microarrays. We have successfully tested the compatibility of the chip with two such scanners, the arrayWoRx (Applied Precision, Issaquah, WA) and the ScanArray 4000 (Packard BioChip Technologies, Billerica, MA), (data not shown).
The inlet structure has been prepared and evaluated in a number of different designs in an effort to ensure compatibility of the chip with ordinary micro pipettes, and with automated liquid handling robots. The inlet of the chip shown in Fig. 2A is equipped with a removable silicone adapter designed to accommodate a standard pipette tip. In comparison the inlet of the chips used in the present work is simply a conical hole with an inner diameter of 300 µm and an opening angle of 60° (cf.Fig. 1C). From the inlet the liquid runs through a very narrow gap, which is designed to protect the hybridization chamber from drying out by ensuring that a minimal liquid surface area is available for evaporation. From here the liquid continues into a ∼2 cm long, 250 µm wide and 100 µm deep channel, which acts as sacrificial buffer if the liquid starts to evaporate during incubation, to delay the liquid meniscus from entering the hybridization chamber.
 | ||
| Fig. 2 (A) The Closed Chip (25 × 76 × 1.1 mm) with a silicone adapter at the inlet (A). The straight hybridization chamber (B) is filled with a dye solution to enhance contrast, since the surface of the chamber is optically transparent and barely visible without the dye. At the end of the hybridization chamber is a hydrophobic stop (C), followed by a meandering waste chamber (D), which retain the liquids used in the assay. The waste chamber is convenient because liquids need only be added but not removed from the chip during an assay. When liquid is introduced into the chip the air inside the chip is allowed to escape through a vent (E) in the distal end of the waste chamber. The circular depressions (F) facilitate proper ejection of the part from the molding cavity. (B) SEM-image of the chip base manufactured by combining injection molding and micro milling. (C) The white frame in B indicates the location of the close-up of a region of the chip where the milled and the molded structures meet. | ||
The chip was designed for single use with the smallest possible volume of target solution as would be appropriate in most biological applications, e.g. when the aim is to measure expression levels in a cDNA sample, genotypes in a PCR-reaction or similar. The dimensions of the hybridization chamber have been carefully optimized to provide as large a surface area as possible for printing of microarrays or immobilization of similar assay components, while at the same time maintaining functional fluidic behaviour and a minimal overall volume. The capillary forces acting on a liquid in the hybridization chamber is larger along the edges of the channel where the liquid is in contact with a surface on 3 sides compared to the central part of the chamber where only the chamber top and bottom adds to the capillary drag. If the chamber is too shallow or too wide the capillary drag will cause “flow shooters” along the edges of the chamber resulting in uneven filling and danger of bubble entrapment during the first filling. Proper dimensions of the hybridization chamber are important for reproducible and trouble-free filling, even when the chamber is filled by pressure rather than by capillary action. Since the hybridization chamber is sealed by a flexible foil, the risk of foil adherence to the bottom of the chamber is also increased, when the ratio of width-to-depth becomes to large. The optimal dimensions of the chamber was found to be 100 µm deep, 2.5 mm wide and 23 mm long resulting in a total volume of the chamber of <10 µL including dead-volume in the channels leading to and from the hybridization chamber. Both top and bottom of the hybridization chamber is carefully designed to have good optical properties. Hence the hybridization taking place in the chamber can be monitored from either side of the chip. In the work presented here the chip is placed with the foil side facing the Peltier element to ensure optimal heat transfer between the Peltier element and the liquid in the hybridization chamber. The detection is performed through the 900 µm thick polymer base in order to avoid the inevitably higher intrinsic fluorescence from the foil compared to the chip base. Accordingly the spots are located on the ceiling of the chamber when the chip is placed in the microscope (cf.Fig. 1C).
In the downstream end of the hybridization chamber there is a capillary stop in the channel leading from the hybridization chamber to the waste chamber. This act to stop the liquid from being dragged into the waste chamber by capillary forces. When the first liquid is loaded the capillary force in the chip will drag the liquid to the capillary stop, even if no external pressure is applied, ensuring complete and reproducible filling of the chip.
The on-chip waste chamber has a volume of ∼140 µL, which enables it to hold liquid from 12–14 fillings of the hybridization chamber, corresponding to 3 assay steps (e.g. one sample loading, a first detection reagent and a second detection reagent) each step followed by washing of the chamber by 3 volumes of buffer. The waste chamber was designed as a meandering channel, rather than a rectangular chamber, to add mechanical stability to the chip and to ensure that the liquid remains a continuous liquid plug to prevent bubble entrapment. The presence of an on-chip waste chamber is convenient during assay handling because liquids need only be added to the chip, but never removed – the introduction of the next assay buffer simply displaces the first into the waste chamber.
In the distal end of the waste chamber there is a small chamber, which is closed by the sealing foil and must be punctured prior to using the chip. The foil can be punctured with any sharp and pointy object like an infusion needle or a scalpel and this puncture then functions as a vent, which allows air to escape from the chip when liquid is introduced.
Sealing of polymer microstructures can be accomplished with several different techniques including simple approaches like gluing or solvent assisted bonding15 and more advanced approaches like ultrasonic welding or infrared laser welding.16 For the chip presented in this article foil sealing was chosen as the preferred method of sealing for several reasons. The use of a thin foil makes it possible to obtain an overall thickness of the chip of 1.1 mm, which is required to ensure compatibility with existing microscope slide handling equipment. If the same overall thickness was to be realized by joining two molded parts (e.g. each with a thickness of 0.55 mm) this would have added tremendously to the complexity of both the design of the microstructures and the molding process of the parts. Furthermore the foil lamination is a very simple process, which is easy to automate and integrate on a production line compared to the alternative methods mentioned above. The process is so simple that we have successfully applied a standard office paper laminator to seal a number of chips. The office laminator is normally used to apply a polymer coating to paper to ensure durability and as such no focus is put on issues like intrinsic fluorescence of the polymer or homogeneous sealing in the micrometer scale. In spite of this we managed to achieve functional sealing of the chips using the lamination foil which accompanied the office laminator. The result was somewhat cruder than the lamination achieved with the dedicated equipment and the fluorescence background was higher, but the results illustrate the simplicity of the technique and holds promise that the technique can even be performed by the end-user of the chip. This would allow users to print their own microarrays in the hybridization chamber of the injection molded base and perform the lamination themselves immediately prior to hybridizing with a target solution.
Polymer requirements
Despite the many advantages of polymers, there are a number of properties that must be considered when choosing an appropriate polymer for a microfluidic chip. The shrinkage factor of the basic polymer (i.e. the final size of the hardened part compared to the dimensions of the mold cavity) is an important parameter, which has to be considered in injection molding applications. If the shrinkage is small (e.g. 0.5%) there are usually no problems, but if the shrinkage is larger (e.g. >1%) this will impose some limitations to the designs that can be realized on the micrometer scale. Different agents are often added to polymers to improve their molding properties e.g. with respect to flow characteristics of the melted polymer, lubrication inside the molding tool or release of the molded part from the tool. Another important property for many lab-on-a-chip applications is the inherent material fluorescence, which frequently does not originate from the polymer material itself, but rather from one or more of the agents that have been added to improve molding properties17 (see section “Background fluorescence” for further discussions).The chip presented in this work was designed to be used with an optical detection system for measurement of fluorescence signals at distinct locations inside the chip, and it was therefore important that the polymer was of high optical quality. Besides a low intrinsic fluorescence we also required a high optical clarity with minimal distortion of the light and a high light transparency in the relevant wavelength range. The chip had to be chemically resistant to solvents commonly used in biological sciences to ensure compatibility with existing protocols and procedures. Furthermore the chip must be able to withstand prolonged exposure to temperatures in the range from 0 °C to 100 °C and preferably also to thermocycling conditions used during PCR. The polymer's propensity to unspecifically adsorb biological macromolecules should also be taken into account, as should its hydrophobicity, which will influence the capillary properties of the micro structures. However these properties can be controlled by subsequent surface modification of the molded parts using a number of different methods.18,19,20,21
The chips were manufactured by injection molding, which produced parts containing some of the functional features. Subsequently, additional structures were added to the molded parts by micro milling. This two step procedure adds the requirement that the molded parts must be machinable by micro milling of structures down to 50 µm. Such structures can be produced directly in the injection molding process, but the two step procedure combines the ability of injection molding to obtain structures of high clarity and low surface roughness, with the flexibility of micro milling to derive an optimal final design of individual structures. The hybridization chamber and the waste chamber are produced by injection molding and the inlet structure, connecting channels and the hydrophobic stop are subsequently micro milled in the molded part. Fig. 2B shows a Scanning Electron Microscope (SEM) image of the hybridization chamber, connecting channels and hydrophobic stop in the final chip. Fig. 2C is a close-up of the region where the molded hybridization chamber and the milled connecting channel meet. The surface of the molded hybridization chamber in the right side of Fig. 2C has a smooth surface compared to the left side, which shows the beginning of the milled connection channel. Due to the surface roughness of milled structures this method is unfit for manufacture of structures for optical detection, because the surface roughness of the structures causes excessive refraction of the light.
The chips were injection molded in polycarbonate (PC), which has very high chemical resistance and thermal stability, a low shrinkage factor and excellent properties for micro milling. The injection molded parts have a transparency of >90% in the wavelength range 350–1100 nm, in the optical path where the material thickness is 900 µm. The molded parts do show some intrinsic fluorescence when compared to a standard glass slide, but it is at an acceptable level for wavelengths above 450 nm. In a mass production scenario where the final design would be produced by injection moulding alone, another polymer might be chosen, which does not meet the requirement for acceptable milling properties.
Background fluorescence
Background fluorescence in the experimental setup can originate from a number of different sources and we have worked to minimize contributions from some of these sources. As mentioned earlier the intrinsic material fluorescence rarely originates from the polymer itself, but is mainly related to additives in the polymer resin. The material autofluorescence therefore has to be balanced against other polymer properties governed by these additives and mainly related to the molding process. Intrinsic fluorescence from parts that are molded or extruded from polymers with a low content of additives, might originate from stress in the material22 or uneven distribution of additives in the molded parts,23 effects which can be avoided or reduced significantly by proper control of the molding process. We have evaluated a number of different polymers including several different types of polycarbonate. Small dummy parts were injection molded and investigated for intrinsic fluorescence until the preferred polymer was located. Using this polymer the injection molding process was then optimized until fluorescence from residual stress was minimized in the molded parts (data not shown).After a microarray was printed in the hybridization chamber of a molded chip base, the chip was closed by lamination with a thin foil. For thin foils the intrinsic fluorescence can often cause problems due to the additives required to extrude very thin foils (e.g. 50–100 µm). Furthermore the foil needs to adhere properly to the chip base, so the foil must be coated with an adhesive, which is either temperature sensitive (so the foil will stick when heated) or pressure sensitive (like ordinary Scotch® tape). These adhesives can result in elevated background fluorescence, but they can also cause other problems like leakage of unwanted substances into the hybridization chamber or they may increase unspecific adsorption of sample molecules, which can then lead to increased background fluorescence. We have investigated a number of heat sensitive and pressure sensitive foils for intrinsic fluorescence, and have found that the heat sensitive foils consistently showed higher fluorescence levels than the pressure sensitive ones at both 530 nm, 595 nm and 685 nm emission wavelengths (data not shown). We ended up using a polyolefinic microplate sealing foil from 3M (9793) and a custom made polypropylene foil.
Passive adsorption of sample molecules to polymers can often be reduced to a minimum by the inclusion of small amounts of detergent in the buffers (e.g. 0.1% Tween®20), which reduce the hydrophobic interactions between the polymer and hydrophobic parts of the biological molecule. Detergent is therefore routinely added to all buffers and passive adsorption is not a significant problem. Furthermore the presence of detergent in the buffers significantly enhances the efficiency of the degassing procedure by reducing the surface tension of the liquid and allowing entrapped gasses to escape more easily. The presence of detergent thus assisted in reducing the risk of bubble formation inside the hybridization chamber under elevated assay temperatures and prolonged hybridizations.
Flow characterization
In microfluidic channels, flow is very often laminar rather than turbulent due to the very low Reynolds numbers. This implies that mixing of joining streams takes place as a result of diffusion only and not by convection. To investigate whether the streams from the two syringe pumps in the experimental setup are properly mixed before entering the hybridization chamber, we performed a mixing experiment with coloured solutions as described in the “Experimental” section. Images of the chip were acquired with different ratios of the coloured and clear solutions running through the hybridization chamber. The colour intensity of an image in a certain position is proportional to the amount of dye at that position and can be taken as a measure of the depth of the structure as described by Broadwell et al.24 Line profiles were acquired perpendicular to the hybridization chamber as illustrated in Fig. 1B. The line profiles from the different conditions addressed during the experiment can be seen in Fig. 3. | ||
| Fig. 3 Profile scans across the width of the hybridization chamber with different ratios of dye and Milli-Q from the two pumps. Initially the chamber is filled with 100% Milli-Q (A, solid), then 100% dye (B, solid), then washed with 100% Milli-Q for 3 min (C, solid), washed with 100% Milli-Q for an additional 2 min (D, dash) and finally filled with 50% dye (E, solid). See text for further details. | ||
First the chip is filled with 100% Milli-Q water resulting in profile A. From the central part of this profile it can be seen that the hybridization chamber itself has no absorbance, relative to the polymer chip base, when filled with a clear liquid. However, along the steep edges of the channel significant absorbance is observed, which is caused by optical refraction of the incident light and should not be interpreted as inhomogeneity of the liquid distribution. The Milli-Q in the chamber is replaced by pumping 100% dyed solution into the chip, resulting in profile B in Fig. 3. This profile reveals an excellent homogeneity of the liquid distributed in the hybridization chamber, when one liquid replaces another. The volume of the hybridization chamber is ∼10 µL and the dead volume of the system from the valve where the liquid streams from the two syringe pumps meet to the point where they enter the hybridization chamber, is approximately 15 µL. To completely replace the liquid in the chamber the pumps must deliver at least 25 µL if no mixing occurs between the injected and the displaced liquids. At 10 µL min−1 for 3 min the pumps have delivered 30 µL and the liquid in the chamber has thus been replaced with only 5 µL surplus. Profile C in Fig. 3 represents washing of the dye filled chamber with 100% Milli-Q for 3 min and illustrates how a reminiscence of the dye can still be seen as a slightly higher background compared to the initial Milli-Q filling (profile A). This background signal indicates that 5 µL surplus (0.5 × the chamber volume) is not enough to wash the hybridization chamber sufficiently. After washing of the chamber with additional 20 µL of Milli-Q (i.e. a total of 2.5 × chamber volume) no trace of the dye is visible (profile D). Finally the chamber is filled with the two pumps pumping simultaneously at 5 µL min−1 each for 3 min, resulting in a mixture of 50% dyed solution at 10 µL min−1. If the liquid streams did not mix properly this would be visible as an inhomogeneous distribution of the dye in the chamber resulting in an uneven profile. From the very even profile E we can thus rule out concentration gradients due to laminar flow conditions on the chip.
Temperature control
To investigate the correlation between the temperature profile programmed into the Peltier controller and the actual temperature inside the hybridization chamber, a micro temperature sensor (probe Ø < 100 µm) was introduced through a hole in the polymer chip into the hybridization chamber (cf.Fig. 1C). This investigation was considered important because of the generally very low heat conductivity of polymer materials. However the very limited thickness of the polymer foil (100 µm) facing the Peltier element and the very small volume of the hybridization chamber (10 µL) result in an excellent correlation between the Peltier temperature and the temperature measured with the micro sensor. The measurement was performed with a constant flow of 1× SSCT-buffer at 10 µL min−1 through the chamber to mimic real assay conditions and a heating and cooling cycle was performed on the Peltier stage raising the temperature to 80 °C and cooling back to 20 °C at a rate of 2 °C min−1. A linear fit to the data from this experiment yielded a correlation of Measured Temperature = 0.96 × Peltier Temperature − 0.65 °C with R2 = 0.999. This corresponds to a maximum deviation of 4 °C at 80 °C between the temperature setting for the Peltier element and the actual temperature measured inside the chamber. The micro sensor has a precision of ±0.5 °C in the range 0–100 °C. When the absolute temperature for carrying out a hybridization reaction is of importance, corrections can be made using this relationship, but for most practical purposes the small inaccuracy can be ignored.Diffusion in microfluidic hybridization assays
Due to the low values of the diffusion coefficients D for the DNA and RNA molecules involved in microarray hybridization assays, the hybridization process turns out to be strongly diffusion limited.To gain insight in the diffusion conditions of microarray hybridizations we have modelled the hybridization of a target T and a capture probe C by the simple reaction T + C ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) CT described by the following coupled diffusion equations:
CT described by the following coupled diffusion equations:
 | (1) |
 | (2) |
In the microarray layout the distance between replicate spots is 4 mm, while the height of the hybridization chamber is 100 µm. To study hybridization to a single spot we therefore solve eqns (1) and (2) for a cylindrical geometry with radius R = 2 mm and height h = 100 µm as illustrated in Fig. 4A. In the center of the bottom plane is placed a spot of radius rs = 50 µm containing the capture probes with an area concentration [C](r,t), where r is the radial coordinate and t is time. The initial area concentration is set to C0 = 10 pmol cm−2 according to previously published data on typical capture probe densities.25,26,27,28
![(A) Illustration of the geometry used in the numerical solution of eqns (1) and (2). The rectangle illustrates the position of the plane for which sectional views are provided in (B). (B) Sectional views illustrating [T] in shades of gray across the hybridization chamber for different times t, where white indicates higher concentration.](/image/article/2004/LC/b311991b/b311991b-f4.gif) | ||
| Fig. 4 (A) Illustration of the geometry used in the numerical solution of eqns (1) and (2). The rectangle illustrates the position of the plane for which sectional views are provided in (B). (B) Sectional views illustrating [T] in shades of gray across the hybridization chamber for different times t, where white indicates higher concentration. | ||
To mimic the properties of a target solution we consider a comparative expression analysis experiment, which is the most common type of experiment that microarrays are used for. In this type of experiment the aim is to measure the relative quantities of different species of mRNA molecules in two samples that are mixed together (one population of mRNA's are labelled with one fluorophore the other population is labelled with another fluorophore). Because the two samples are mixed and hybridized together diffusion will not affect the relative signals from the two different mRNA populations, but diffusion might affect the detection limit of the analysis. Following an amplification step the average size of the target molecules (complementary DNA, cDNA or amplified RNA, aRNA) will be in the range of 800 bases7 having a molecular weight of 250 kDa and an estimated diffusion coefficient D = 4 × 10−8 cm2 s−1.29 The volume concentration of the target molecule in question is denoted [T](r,z,t), where z is the vertical coordinate, and the initial target concentration is set to T0 = 10 pM corresponding to mRNA-levels from a medium expressed gene.5 For the rate constants we applied values previously reported for hybridization of DNA target to an immobilized 15-mer capture probe using the BIAcore technique;30k1 = 12 × 103 M−1 s−1 and k2 = 2.9 × 10−4 s−1.
From the model we obtain profiles through the hybridization volume showing [T] at different locations in the hybridization chamber for a given time t as illustrated in Fig. 4B. In this model the hybridization rate given by k1 drives the target concentration at the surface of the spot [T](0,t) towards zero, much faster than the diffusion process can refuel the hybridization by bringing in new target molecules. Thus a concentration front is created where [T]
= T0, which is slowly moving away from the spot as illustrated by the [T]-profiles for different t's in Fig. 4B. As expected the position r0 of the receding front where [T]
= T0 is found to be given by r0(t)
≈
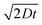 . In addition to the numerical simulations we have found an analytical solution to eqns (1) and (2) in one spatial dimension, for a suitable set of boundary conditions. This solution along with details of the actual numerical work will be described elsewhere.31
. In addition to the numerical simulations we have found an analytical solution to eqns (1) and (2) in one spatial dimension, for a suitable set of boundary conditions. This solution along with details of the actual numerical work will be described elsewhere.31
A qualitative estimate of the diffusion phenomena can also be obtained by estimating the root mean square distance travelled by a molecule of a given size (i.e. with a given D) in the time span of an experiment by r
=
.32 For an 800-mer in a 24 h experiment this calculation yields r800,24h
≈ 800 µm and similar calculations for a 5000-mer with D
= 1 × 10−8 cm2 s−1 and a 30-mer with D
= 40 × 10−8 cm2 s−1 yields r5000,24h
≈ 400 µm and r30,24h
≈ 3.7 mm respectively. For the predominant 800-mer oligonucleotide any given capture probe thus samples a volume of only (800 µm)3
≈ 500 nL during the entire experiment. Since this is considerably less than both the usual sample volumes of 25–200 µL (obtained when using a microscope slide with a cover slip) and the <10 µL sample volume of the microfluidic chip, the overall number of target molecules in the sample in not the limiting factor in any of the assays. Thus we do not expect the detection limit of the assay to be affected by scaling down.
In an effort to minimize the dependence of the hybridization reaction on diffusion during kinetic measurements, we applied a constant flow of a concentrated target solution, with the velocity field u, through the hybridization chamber. A flux [T]u is thus sent passed the spot and this changes the pure diffusion process described in eqn (1) into a diffusion-convection process know as Taylor dispersion.33 The presence of u leads to the following modification of eqn (1):
 | (3) |
The numerical solution of eqns (2) and (3) show that indeed one can speed up the hybridization process by flushing sample through the hybridization chamber. In a particular calculation we have calculated an increase in [CT] by a factor of 3 by increasing the velocity from u
= 0 mm s−1 to u
= 1 mm s−1
(= 15 µL min−1 for the current dimensions of the hybridization chamber). The effect of the flow velocity is to move the front of the [T]
= T0 concentration level closer to the spot. In fact, a sufficiently high flow rate will prevent the -movement of the front and actually keep [T]
= T0 stationary at the spot surface.
In conclusion, since the majority of the molecules in a biologically relevant target solution have very slow diffusion rates, diffusion is the detection limiting factor in both standard microarray experiments and microfluidic devices unless stirring of the sample is applied. Since most microfluidic devices have volumes larger than the assay volume interrogated by a single capture probe, the detection limit of an assay will not be affected by miniaturization. However, for low abundant target molecules in very small micro(nano)fluidic devices the absolute number of molecules available for detection might become a limiting factor unless the target solution is pre-concentrated. In situations where it is feasible to increase the concentration of the target solution, this will on the other hand have a direct positive influence on the apparent detection limit of the assay. The results also illustrate the importance of using a proper array layout where replicate spots are located sufficiently far apart as it is done in the arrays used in this work. If replicate spots are printed closer to each other than r0 (where [T] = T0) they will compete for the same target molecules and effectively decrease the detection limit. Flushing or stirring of the sample will increase both the hybridization rate and the assay detection limit by suppressing diffusion limitations and increasing the effective volume interrogated by individual capture probes.
On-line hybridization measurements
As described in the Experimental section an array of capture probes were spotted in the hybridization chamber. A total of 54 different oligos were synthesized consisting of different capture sequences some of which contained Locked Nucleic Acids (LNA). Some capture probes were synthesized with different spacers and some were spotted in the array at different probe concentrations. All these variations resulted in an array of 128 spots, which was replicated 4 times inside the hybridization chamber. By hybridization with a solution containing a single target it was possible to obtain on-line hybridization information from 4 × 128 different capture probe spots. Large amounts of data were acquired in this way by hybridizing with different targets under different conditions. These data will form the basis of a thorough investigation of the properties of Locked Nucleic Acids in surface hybridizations to be published elsewhere.To evaluate the on-line measurement capability of the chip and the setup, samples of the data are presented in Fig. 5 illustrating various aspects of the chip and setup performance.
 | ||
| Fig. 5 (A) This figure illustrates the effect of flushing target solution through the chamber versus a hybridization without flow where diffusion quickly becomes the limiting factor. (B) Demonstrates the reproducibility between two identical hybridizations to the same chip, separated by a cleaning of the chip by flushing with buffer at elevated temperature. (C) Spot-to-spot variation between replicates in the same chip is indicated by the error-bars representing the standard deviation. From this a similar hybridization and wash curves, valuable information can be derived about on and off rates of the hybridization reaction. | ||
In Fig. 5A the effect of flushing the chamber during hybridization is illustrated. As it appears, flushing the chamber with fresh target solution and thus keeping the target concentration constant, does not affect the initial hybridization rate. This indicates that the initial concentration of the 30-mer target used in these experiments [T] = 0.01 µM is sufficiently high to supply the immobilized capture probes with target molecules at the current rate of hybridization. After 30 min however it is obvious that the hybridization without flow, which is dependent on diffusion, is lagging behind because the target solution in the vicinity of the capture probe is depleted and diffusion is too slow to supply new target molecules. Further increasing the flow rate from 5 µL min−1 to 10 µL min−1 did not increase the rate of hybridization any further.
Fig. 5B demonstrates the excellent reproducibility obtained when the same chip is hybridized twice with the same target solution. Between the two on hybridizations shown in this figure the chip was regenerated by washing with pure buffer at elevated temperature, thus stripping all target molecules from the capture probes. Interestingly the two repetitions in the figure are actually the second and the third hybridization on the same chip rather than the first and second. Comparing the first and the second hybridization would yield a result similar to Fig. 5A with the open circles representing the first hybridization, which has turned out to be somewhat slower than the subsequent hybridizations. If a chip is treated with a buffer wash at elevated temperature prior to use, the first hybridization does not appear to be slow indicating that the wash procedure activates the immobilized capture probes. This is probably caused by washing off capture probes that are not covalently immobilized but retained in the spot by probe–probe hybridizations, thus occupying sites for target hybridization.
The hybridization in Fig. 5C illustrates a complete hybridization experiment with the first 60 min representing the end of a wash cycle and the subsequent 300 min representing a hybridization in which the target concentration is kept constant at [T] = 0.01 µM. The period from t = 360 min to t = 720 min represent the washing under which the chamber is flushed with pure buffer of the same composition as used during hybridization. From this curve and several others like it it is possible to derive valuable information about the hybridization process. The error bars on the curve in this figure represent the standard deviation within 4 replicate spots in the hybridization chamber. The larger error bars at t = 300 min and t = 500 min are probably caused by a passing bubble or fluorescent particle, which is sampled together with one of the 4 replicate spots. The experimental setup allows some parts of the experiment to be sampled at a higher rate than others, illustrated in this figure by the more closely packed data point in the parts of the curve where the slope is expected to be highest.
Conclusion
In this article we have presented a very mature design of a disposable single use hybridization chamber manufactured by injection molding and foil lamination. This approach yields parts of very high quality and tight tolerances. The design has been optimized for mass production without compromising the fluidic behaviour or optical properties of the chip. We have demonstrated the performance of the chip with respect to homogeneous liquid distribution and the ability to reliably perform a DNA:DNA hybridization. The chip was applied in a flexible experimental setup which provided on-line measurements and enabled close control of the assay conditions and the ability to gradually change these during the experiment.The chip and the experimental setup presented in this work will enable powerful and exciting new experiments that are impossible to perform using standard open faced microarrays. Kinetic and thermodynamic measurements of nucleotide hybridizations have traditionally been performed in solution, and existing prediction algorithms and theoretical models are all based on these measurements. These algorithms are therefore not necessarily valid for solid phase hybridizations and the ability of existing theories to model the hybridization of a target to an immobilized capture probe on a solid surface is not yet fully investigated. The setup and polymer microfluidic chip presented in this article will enable the highly parallel measurements of array based hybridizations needed to develop a strong theoretical understanding of solid phase hybridizations.
The use of on-line measurements of hybridization events further enable a number of experiments that are impossible to perform using only end-point measurements. In microarray hybridization experiments it is normally desirable that all capture probes in the microarray have approximately the same Tm in order for them to be able to hybridize to their target under the same experimental conditions. The use of a closed chip for microarray hybridization experiments eliminates this requirement because the chip can be hybridized at several different stringency conditions sequentially. This can e.g. be achieved by hybridizing the chip at low temperature and then heating the chip while recording the melting profiles for all the capture probes simultaneously. Stringency could also be changed by applying different buffer compositions during a washing step. Alternatively the chip could be used to follow the signal build-up from all capture probes in a standard expression profiling microarray experiment. This would reveal at what time the signals from individual capture probes reach equilibrium, and it would be possible to monitor for any unpredicted kinetic behaviour of the hybridization events. It could also find use in other applications where control of the hybridization parameters are important.
It has previously been reported that Locked Nucleic Acids (LNA) significantly enhances the affinity for a complementary DNA strand, enabling the use of shorter probes and thereby increasing the discriminatory power.34,35,36 The hybridization properties of LNA are not yet fully investigated and the chip will be useful in further investigations of the kinetic properties of LNA or novel nucleotide analogues, by enabling highly parallel experiments that can address multiple capture probes simultaneously.
If applied, the chip will be able to make a lot of existing assays easier to perform by eliminating some of the difficult handling steps. Most existing methods and protocols for microarray hybridization are based on open-faced arrays where hybridizations are performed under a coverslip and washing is performed by soaking the substrate in a buffer trough. These methods are labour intensive, require skillful handling and are very difficult to automate. The chip presented in this work is developed to be compatible with automation and robot handling and to eliminate the difficult handling steps connected to open-faced microarrays.
Acknowledgements
The chip was designed in collaboration with STEAG microParts (SmP), Dortmund, Germany, who has also manufactured the chip base by injection molding. Subsequent to printing of microarrays in the hybridization chamber the chips were sealed by foil lamination by SmP. SEM images and profilometry measurements were performed with kind assistance by Dorte Iven Larsen at the Center for Microtechnology and Surface Analysis, Danish Technological Institute, Taastrup, Denmark. The work has been partially funded by The Danish Academy for Technical Sciences (ATV) through a PhD fellowship.References
- S. C. Terry, J. H. Jerman and J. B. Angell, A gas chromatographic air analyzer fabricated on silicon wafer, IEEE Transactions on Electron Devices, 1979, ED-26(12), 1880–1886 Search PubMed.
- A. Manz, N. Graber and H. M. Widmer, Miniaturized total chemical analyses systems. A novel concept for chemical sensing, Sens. Actuators, B, 1990, B1(1), 244 CrossRef.
- E. M. Southern, U. Maskos and J. K. Elder, Analyzing and comparing nucleic acid sequences by hybridization to arrays of oligonucleotides: evaluation using experimental models, Genomics, 1992, 13(4), 1008–1017 CAS.
- S. P. Fodor, R. P. Rava, X. C. Huang, A. C. Pease, C. P. Holmes and C. L. Adams, Multiplexed biochemical assays with biological chips, Nature, 1993, 364(6437), 555–556 CrossRef CAS.
- R. Ramakrishnan, D. Dorris, A. Lublinsky, A. Nguyen, M. Domanus, A. Prokhorova, L. Gieser, E. Touma, R. Lockner, M. Tata, X. Zhu, M. Patterson, R. Shippy, T. J. Sendera and A. Mazumder, An assessment of motorola codelink microarray performance for gene expression profiling applications, Nucleic Acids Res., 2002, 30(7), e30 CrossRef.
- P. Stafford, T. Sendera, T. Kaysser-Kranich and C. Palaniappan, High-quality microarray data using codelink bioarray platform, Life Sci. News, 2003, 13, 3–5 Search PubMed.
- DNA Microarrays: A Molecular Cloning Manual, ed. David Bowtell and Joseph Sambrook, Cold Spring Harbor Laboratory Press, New York, 2003 Search PubMed.
- Y. Liu and C. B. Rauch, DNA probe attachment on plastic surfaces and microfluidic hybridization array channel devices with sample oscillation, Anal. Biochem., 2003, 317(1), 76–84 CrossRef CAS.
- C. Peter, M. Meusel, F. Grawe, A. Katerkamp, K. Cammann and T. Borchers, Optical DNA-sensor chip for real-time detection of hybridization events, Fresenius J. Anal. Chem., 2001, 371(2), 120–127 CrossRef CAS.
- Y. Wang, B. Vaidya, H. D. Farquar, W. Stryjewski, R. P. Hammer, R. L. McCarley, S. A. Soper, Y. W. Cheng and F. Barany, Microarrays assembled in microfluidic chips fabricated from poly(methyl methacrylate) for the detection of low-abundant DNA mutations, Anal. Chem., 2003, 75(5), 1130–1140 CrossRef CAS.
- R. Lenigk, R. H. Liu, M. Athavale, Z. Chen, D. Ganser, J. Yang, C. Rauch, Y. Liu, B. Chan, H. Yu, M. Ray, R. Marrero and P. Grodzinski, Plastic biochannel hybridization devices: A new concept for microfluidic DNA arrays, Anal. Biochem., 2002, 311(1), 40–49 CrossRef CAS.
- M. D. Matteuci and M. H. Caruthers, Synthesis of deoxyoligonucleotides on a polymer support, J. Am. Chem. Soc., 1981, 103, 3185–3191 CrossRef CAS.
- A. A. Koshkin, S. K. Singh, P. Nielsen, V. K. Rajwanshi, R. Kumar, M. Meldgaard, C. E. Olsen and J. Wengel, LNA (locked nucleic acids): Synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition, Tetrahedron, 1998, 54(14), 3607–3630 CrossRef CAS.
- T. Koch, N. Jacobsen, J. Fensholdt, U. Boas, M. Fenger and M. H. Jakobsen, Photochemical immobilization of anthraquinone conjugated oligonucleotides and PCR amplicons on solid surfaces, Bioconjug. Chem., 2000, 11(4), 474–483 CrossRef CAS.
- H. Klank, J. P. Kutter and O. Geschke, CO2-laser micromachining and back-end processing for rapid production of PMMA-based microfluidic systems, Lab Chip, 2002, 2(4), 242–246 RSC.
- Leister, Novolas laser systems, http://www.novolas.com/, [Accessed November 2003] 2003.
- A. J. Bur and S. C. Roth, Fluorescence based temperature measurements and applications to real-time polymer processing, in Society of Plastics Engineers Technical Conference, Society of Plastics Engineers, Orlando, FL, 2000, vol. 2, p. 348 Search PubMed.
- P. Nahar, N. M. Wali and R. P. Gandhi, Light-induced activation of an inert surface for covalent immobilization of a protein ligand, Anal. Biochem., 2001, 294(2), 148–153 CrossRef CAS.
- A. C. Henry, T. J. Tutt, M. Galloway, Y. Y. Davidson, C. S. McWhorter, S. A. Soper and R. L. McCarley, Surface modification of poly(methyl methacrylate) used in the fabrication of microanalytical devices, Anal. Chem., 2000, 72(21), 5331–5337 CrossRef CAS.
- C.-M. Chan, T.-M. Ko and H. Hiraoka, Polymer surface modification by plasmas and photons, Surf. Sci. Rep., 1996, 24, 1–54 CrossRef CAS.
- M. H. Jakobsen and T. Koch, Method of photochemical immobilization of ligands using quinones, PCT, WO 96/31557, 1996.
- Taiji Ikawa, Tohru Shiga and Akane Okada, Measurement of residual stresses in injection-molded polymer parts by time-resolved fluorescence, J. Appl. Polym. Sci., 2002, 83(12), 2600–2603 CrossRef CAS.
- J. A. Martins, J. Seixas, M. J. Oliveira, A. Maio and A. S. Pouzada, Optical properties of injection-molded polystyrene scintillators. ii. Distribution of dopants, J. Appl. Polym. Sci., 2003, 88(11), 2714–2718 CrossRef CAS.
- I. Broadwell, P. D. I. Fletcher, S. J. Haswell, T. McCreedy and X. L. Zhang, Quantitative 3-dimensional profiling of channel networks within transparent ‘lab-on-a-chip’ microreactors using a digital imaging method, Lab Chip, 2001, 1, 66–71 RSC.
- A. W. Peterson, R. J. Heaton and R. M. Georgiadis, The effect of surface probe density on DNA hybridization, Nucleic Acids Res., 2001, 29(24), 5163–5168 CrossRef CAS.
- A. Vainrub and B. M. Pettitt, Coulomb blockage of hybridization in two-dimensional DNA arrays, Phys. Rev. E, 2002, 66, 41905 CrossRef.
- A. B. Steel, R. L. Levicky, T. M. Herne and M. J. Tarlov, Immobilization of nucleic acids at solid surfaces: Effect of oligonucleotide length on layer assembly, Biophys. J., 2000, 79(2), 975–981 CAS.
- F. Fixe, M. Dufa, P. Telleman and C. B. V. Christensen, Functionalization of poly-(methyl methacrylate) (PMMA) as a substrate for DNA microarrays, Nucleic Acids Res., 2003, in press.
- G. L. Lukacs, P. Haggie, O. Seksek, D. Lechardeur, N. Freedman and A. S. Verkman, Size-dependent DNA mobility in cytoplasm and nucleus, J. Biol. Chem., 2000, 275(3), 1625–9 CrossRef CAS.
- K. K. Jensen, H. Orum, P. E. Nielsen and B. Norden, Kinetics for hybridization of peptide nucleic acids (PNA) with DNA and RNA studied with the BIAcore technique, Biochemistry, 1997, 36(16), 5072–5077 CrossRef CAS.
- H. Bruus and M. Noerholm, Manuscript in preparation: Diffusion in microfluidic hybridization assays, 2004.
- P. W. Atkins, Physical Chemistry, Oxford University Press, Oxford, 4th edn., 1990 Search PubMed.
- R. F. Probstein, Physiochemical Hydrodynamics, An Introduction, John Wiley & Sons, New York, 2nd edn., 1994 Search PubMed.
- H. Orum, M. H. Jakobsen, T. Koch, J. Vuust and M. B. Borre, Detection of the factor V Leiden mutation by direct allele-specific hybridization of PCR amplicons to photoimmobilized locked nucleic acids, Clin. Chem., 1999, 45(11), 1898–1905 CAS.
- N. Jacobsen, J. Bentzen, M. Meldgaard, M. H. Jakobsen, M. Fenger, S. Kauppinen and J. Skouv, LNA-enhanced detection of single nucleotide polymorphisms in the apolipoprotein E, Nucleic Acids Res., 2002, 30(19), e100 CrossRef.
- P. Mouritzen, A. T. Nielsen, H. M. Pfundheller, Y. Choleva, L. Kongsbak and S. Moller, Single nucleotide polymorphism genotyping using locked nucleic acid (LNA), Expert. Rev. Mol. Diagn., 2003, 3(1), 27–38 Search PubMed.
| This journal is © The Royal Society of Chemistry 2004 |