Integrated polymer chip for two-dimensional capillary gel electrophoresis
Anja
Griebel
,
Sabine
Rund†
,
Friedhelm
Schönfeld
,
Wolfgang
Dörner‡
,
Renate
Konrad§
and
Steffen
Hardt
*
Fluidics & Simulation Department, Institut für Mikrotechnik Mainz GmbH – IMM, Carl-Zeiss-Str. 18-20, 55129 Mainz, Germany. E-mail: hardt@imm-mainz.de; Fax: +49 (0) 6131 990 205
First published on 13th November 2003
Abstract
Two-dimensional (2D) gel electrophoresis (GE) is one of the most powerful methods for nucleic acid and protein separation, but generally suffers from high laboratory efforts connected with high analysis costs. Therefore, we herein present the development of a miniaturized 2D capillary GE (CGE) device which allows for an efficient protein separation in analysis times of about 1.5 h. This integrated 2D-CGE chip comprises a first channel for isoelectric focussing (IEF), a second specially designed transfer channel, 300 parallel micro channels, each having a cross section of 50 µm × 50 µm, and buffer reservoirs. The present work discusses fabrication aspects, in particular the combination of different microfabrication technologies, experimental separation performances of isoelectric focussing (IEF) and CGE, and presents computer simulations and first experimental results of protein transfer from the first to the second dimension.
Introduction
Two-dimensional (2D) analysis remains one of the most frequently applied methods due to its ability to separate complex mixtures of proteins and to follow multigenic phenomena at the level of whole cell tissues and even whole organisms. Besides relatively high technical effort and lack of inter-laboratory reproducibility of 2D electrophoresis (2DE), long analysis times and insufficient software to evaluate 2D images on a computer are the main reasons for this method not being a routine method for fast characterization of protein mixtures today.1 The above mentioned drawbacks have been addressed by various concepts: standardization of the conditions of 2DE experiments, automation and advanced data processing. The usage of precast gels, simultaneous separation of identical protein mixtures in one 2DE experiment known as 2D differential gel electrophoresis (2D DIGE)2 or development of partially automated 2D electrophoresis apparatus (e.g. Phast-System, Pharmacia) are mentioned as well as development of more sophisticated computer based data processing on the level of initial spot recognition,3 comparison of the expression of particular proteins on several gels, and comparison of databases created in different laboratories.3Alternatively, miniaturization opens up a route for improvements in 2DE performance.4 The well-known capillary gel electrophoresis (CGE) allows rapid sample separation in comparison to slab gels5 and can be performed at high electric field, due to the efficient removal of Joule heat. Another advantage of CGE is the possibility of real-time detection. However, there are some challenges connected to the use of CGE for miniaturized 2DE, among others the realisation of the transfer of proteins from the first to the second dimension and a highly parallel separation. Chen et al.4 presented a method for carrying out 2D-CGE. They realized the transfer from the first to the second dimension by means of a poly(dimethylsiloxane) (PDMS) structure, which allows for reversible connections between different devices. First results showed the functionality of such a system. However, the resolution of this prototype for 2D-CGE was relatively low, since a comparatively small number of capillaries for the second dimension (100 capillaries, cross section 100 µm × 100 µm) were used.
Generally, a modular system with reversible connections prevents or at least complicates efficient process automation. To this end, an alternative integrated approach is chosen by the authors for the development of a disposable 2D-CGE polymer chip.
Experimental section
Chip design
The developed microstructured electrophoresis chip combines the advantages of immobilized pH gradients (IPG), such as high resolution and long supportability in the first dimension with the speed of highly parallel sodium dodecylsulfate (SDS)-CGE in the second dimension. The credit-card sized chip (85 mm × 55 mm × 2 mm) contains a cavity for a special IPG strip on the top side, furthermore, two buffer reservoirs for the electrodes used for the SDS-CGE, cf.Fig. 1. Both reservoirs are connected via 300 parallel channels (50 µm × 50 µm cross section, 64 mm long). The IPG-strip cavity and the 300 parallel channels are arranged perpendicular to each other and are physically connected by an opening of 50 µm (Fig. 1). After IEF separation, electrodes are inserted into the IEF cavity and the buffer reservoirs. The T-like electrode configuration allows realizing the protein transfer without tailing. The SDS-CGE is driven by the two electrodes placed in the buffer reservoirs. Details concerning electrode configuration and switching are given in a following section.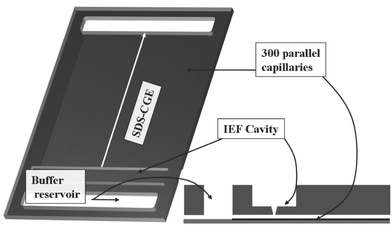 | ||
| Fig. 1 PMMA chip for 2DE. The credit-card sized chip (85 mm × 55 mm × 2 mm) contains a cavity for IEF and 2 buffer reservoirs for electrodes, respectively. These buffer reservoirs are physically connected by 300 parallel channels (50 µm × 50 µm cross section, 64 mm long) for SDS-CGE. The cavity for IEF and the CGE channels are connected to each other by an opening of 50 µm width (encircled region, see also Fig. 10). | ||
Device fabrication
The chip is built up of two different parts, a microstructured polymer plate with 300 open channels and a cover containing the buffer reservoirs and the IEF cavity with the connection gap (cf.Fig. 1). In that way different fabrication methods can be utilized according to the particular needs. The 300 micro channels were realized by hot embossing of 250 µm PMMA sheets (Plexiglas® Röhm, Darmstadt, Germany),6 or by injection moulding which turned out to be most appropriate for high quality replication of the microstructures. To this end a nickel mould insert was made by the following steps: silicon was pattered by UV lithography and structured by Advanced Silicon Etching7,8 (ASE™, Multiplex ICP Etching System, Surface Technology Systems plc, Newport, UK), an anisotropic deep etching process which enables etching of silicon with high aspect ratios (up to 15:1) and nearly perpendicular sidewalls (89° ± 1°). After deposition of a metal seed layer, electroforming9 of nickel and removal of silicon by wet etching, the master structure for the polymer moulding process was obtained (Fig. 2). | ||
| Fig. 2 Nickel mould insert with microstructures. A silicon structure patterned by ASE™ serves as master. After deposition of a metal seed layer and electroplating of nickel, the silicon was removed resulting in a mould insert. | ||
The PMMA cover plate comprising the two buffer reservoirs, the cavity for IEF (all 6 mm × 46 mm in lateral dimension) and the connection between the two dimensions (50 µm × 46 mm) were produced by mechanical micro machining.
After cleaning with isopropanol and O2-plasma treatment (Tegal Corp. Plasmaline 515, USA) both parts were irreversibly connected by solvent bonding. An abraded glass plate was covered by a thin layer of isopentylacetate using a spin coater (CPII; Convac, Germany) at 1000 rpm for 2 s. This wetted glass plate serves as a stamp-pad for solvent application onto the structured side of the PMMA foil. Afterwards the wetted, microstructured PMMA foil was aligned with the cover plate and tightly pressed onto it for 10 min. As can be seen in Fig. 3 by this particularly adapted bonding protocol a tight coverage is achieved without damaging of the microstructures.
 | ||
| Fig. 3 Electron micrograph showing a cut through the 2DE chip. The microstructured PMMA foil (250 µm thickness) shown at the bottom is covered with a PMMA plate by solvent bonding. | ||
In order to suppress electroosmotic flow, a tight linkage between the gel and the channel walls is needed. To this end the channel walls were treated by Serva Electrophoresis GmbH (Heidelberg, Germany) based on procedures described in two patents.10,11
Reagents
Immobiline, ReadySol IEF (40% T, 3% C), and IPG buffer pH 3–10 were purchased from Amersham Biosciences Europe GmbH (Freiburg, Germany). Pullulan was purchased from Polysciences Europe (Eppelheim, Germany). Rotiphorese® NF-Urea, sodium dodecylsulfate (SDS), and bromophenol blue were received from Carl Roth GmbH & Co (Karlsruhe, Gemany). 3-[(3-Cholamidopropyl)-dimethylammonio]-propanesulfonate (CHAPS), tris(hydoxymethyl)aminomethane (Tris) and myoglobin from horse, were purchased from Sigma-Aldrich Chemie GmbH (Taufkirchen, Germany). 2-(Cyclohexylamino)-ethanesulfonic acid (CHES), agarose without electroendosmosis (EEO), dithiothreitol (DTT), Serva Violett 17, and iodoacetamide were obtained from Serva Electrophoresis GmbH (Heidelberg, Gemany). Alexa Fluor 488 protein labeling kits were purchased from Molecular Probes Europe BV (Leiden, The Netherlands).Preparation of IPG strips
The IPG gels (4% and 6% acrylamide), were prepared according to Rigetti12,13 and Görg et al.14 with the following changes. ReadySol IEF solution was used as acrylamide solution. The size of the casting cassette was 60 mm × 170 mm. Since a permeable IPG-strip support structure is required for the protein transfer from the first to the second dimension, a polyester net (ServaNet from Serva Electrophoresis GmbH, Heidelberg, Germany) was used as a support structure instead of a GelBond PAG film commonly used in commercially available IPG strips. The total volume of the gel polymerization solution was 3.9 ml used to obtain a gel with dimensions of 40 mm × 150 mm × 0.5 mm. After drying, the IPG gel was cut into strips (3 mm × 44 mm) using a paper guillotine. Then the strip was placed in a special chamber, a corresponding laboratory setup is shown in Fig. 4. This chamber with the IPG strip can be prepared ready-to-use and stored for several months at −20 °C. The quality of the IPG strips can be quickly checked by rehydrating at 22–25 °C in 1 ml solution of 8 M urea, 2 % (w/v) CHAPS, 0.0002 % (w/v) bromophenol blue, 10 mM DTT and 20 µg IEF marker (Serva Electrophoresis GmbH, Heidelberg, Germany) for 1.5 h. IEF was carried out at 20 °C over a period of 20 min with electrical field strengths of 170 Vcm−1 at the start and 860 Vcm−1 at the end of the run. Staining of proteins with Serva Violet 17 solution was done according to the standard protocol of Serva Electrophoresis GmbH. The protocol revealed a good separation performance of the different IEF marker proteins.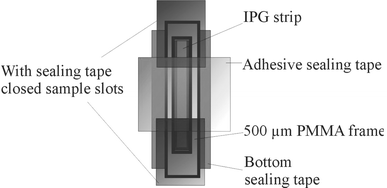 | ||
| Fig. 4 Storage and rehydration cassette (laboratory setup) for IPG strips. The IPG strip is contained in a PMMA frame (inner dimensions: 5 × 45 mm) with adhesive sealing tape of polyolefine (Nalge Nunc International Corporation, Rochester, USA) used as bottom and top sealing. For a bubble-free filling with 140 µl rehydration buffer the sealing tape at the ends is removed. For rehydration the cassette is sealed again. | ||
Preparation of the separation matrix for SDS-CGE and filling of the channels
For CGE the following solution (sieving matrix) was used: 15 % (w/v) pullulan,15 0.1 M CHES, 0.1 M Tris-Cl, 0.1% (w/v) SDS pH 8.8. The solution was heated up to 50 °C for 2–4 h and heavily stirred several times. The separation matrix was filled into the channels of the chip by application of vacuum at room temperature. The 50 µm connection nut was filled with 10 µl of hot stacking gel solution of 0.5% agarose in 0.5 M Tris-Cl pH 6.8.Isoelectric focussing
IPG strips stored at −20 °C in the above mentioned repository were heated up to room temperature. Both sample slots were unsealed and 0.25–40 µg marker protein in 140 µl rehydration buffer (8 M urea, 2 % (w/v) CHAPS, traces of bromophenol blue, and 10 mM DTT was pipetted into one window of the IPG chamber. Both IPG chamber windows were closed with adhesive sealing tape of polyolefine (Nalge Nunc International Corporation, Rochester, USA). Strips were rehydrated for 16–20 h at room temperature. Following rehydration the adhesive tape from the sample windows was removed, and electrodes were placed on filter pads, wetted with deionized water, at each end of the IPG strip. IEF was performed at 20 °C in three steps: (a) 100 V for 15 min, (b) 1000 V for 15 min, (c) 2000 V for 15 or 30 min, with an automated power supply control with a maximum current of 50 µA per strip.After isoelectric focussing, the adhesive tape of the top from IPG chamber was removed, and the IPG strips were stained with Serva Violett 17 according to a standard protocol of Serva Electrophoresis GmbH or prepared by two equilibrations steps (first: 1% DTT in 100 mM CHES, 100 mM Tris-Cl, 2% (w/v) SDS; second: 2.5% jodoacetamide in 100 mM CHES, 100 mM Tris-Cl, 2% (w/v) SDS, 3 min with 150 µl each) prior to transfer to the second dimension, i.e. SDS-CGE.
SDS-CGE
First a rehydrated IPG strip was inserted in the corresponding cavity. Alternatively, 10 µl of a denaturated protein solution or protein mixture in a hot stacking gel solution of 0.5 % agarose in 0.5 M Tris-Cl pH 6.8 were filled into the 50 µm connection nut between the first and the second dimension of the chip. Electrodes were placed in the IEF cavity and in the anodic buffer reservoir (cf.Fig. 1). In the IEF cavity a solution of 400 µl 100 mM CHES, 100 mM Tris-Cl, 0.1 % (w/v) SDS was added and the anodic reservoir was filled with the sieving matrix precursor. Transfer and SDS-CGE were performed at 8 °C at a maximum voltage and current of 2 kV and 1 mA, respectively.Labeling of marker proteins
Marker proteins were labeled with Alexa Fluor® 488 protein labeling kit according to the standard protocol.Instrumental setup
A laboratory setup of an instrument was developed in co-operation with Herolab GmbH (Wiesloch, Germany). It consists of programmable power supplies (HCN 35–3500; HCN 35–6500, HCB 14–1250 all FuG Electronic GmbH, Rosenheim, Germany) with electrodes of glassy carbon (Sigradur G®, HTW GmbH, Thierhaupten, Germany), a CCD detection line (type SK 5150, DJR series, 30 MHz sensor 36 mm, 5150 pixel arranged in a line), a lens (Sigma 50 mm, F 1:2,8), a water cooling unit, and a 25 mW Ar-ion-laser (LG-Laser Technologies GmbH, Kleinostheim, Germany) as fluorescence-inducing light source. The CCD detection line is arranged orthogonal to the micro channels of the chip and its position can be varied along the CE separation length. The power supplies and the CCD detection line are controlled by software developed at Herolab.Results and discussion
IEF
For performance evaluation, IEF was carried out on 40 mm IPG strips (44 mm including reinforced ends) within the disposable cassette shown in Fig. 4. The cassette does not require a mineral oil overlay and the strips are rehydrated directly in the cassette. By using the prepared cassettes, IEF experiments are greatly simplified. After the cassette is warmed up to room temperature, the adhesive tape has to be removed from the sample slots and the sample containing rehydration solution has to be filled into the chamber. The actual IEF experiment without rehydration takes only between 45 to 60 min compared to 2–8 h needed to focus proteins in a conventional oil-immersion system. Besides the simplified handling, the reproducibility of the IEF results is increased as well.Fig. 5 shows an IEF result for Alexa 488 labelled myoglobin. The labelled protein allows for monitoring the efficiency of protein transfer by optical inspection, a prerequisite for further analysis and optimisation. For visualization without any further optics large amounts of protein (5–20 µg) were used. The actual transfer of isoelectrically focussed protein into the 50 µm wide capillaries requires much lower concentrations to avoid an overloading of the capillaries. Sample amounts down to 0.25 µg were tested, less should be possible.
 | ||
| Fig. 5 IEF of Alexa 488 labeled myoglobin. The IPG strip (pH 4–9) was rehydrated for 17 h at room temperature with 5 µg protein in 140 µl rehydration solution (8 M urea, 2 % (w/v) CHAPS, 0.0002 % (w/v) bromophenol blue, 10 mM DTT) visualized with a blue transilluminator (Herolab GmbH, Wiesloch, Germany) and a 550 nm filter. The additional bands may result from carbamylation during rehydration. The IEF was carried out at 20 °C using a power supply protocol with a 50 µA limit per strip and the following steps: (a) 100 V for 15 min, (b) 1 kV for 15 min, (c) 2 kV for 30 min. The amount of protein normally can be reduced down to 0.25 µg or less. | ||
Due to the possibility of preparing certain pH ranges between pH 4 and 9 according to the sample investigated, IPG strips are a flexible tool in 2D electrophoresis for high resolution separation. If IEF is carried out on the chip, the permeable support structure can serve as a barrier between the two separation dimensions. However, when applying appropriate voltages it enables protein migration into the micro channels.
SDS-CGE
Fig. 6 shows a separation result of a SDS-CGE experiment obtained for a mixture of six proteins (Fluorescent High Molecular Weight Standard, Sigma, Taufkirchen, Germany). For these studies the protein mixture was directly applied to the stacking gel filling the connection nut between the IEF cavity and the micro channels. In the first experiments a mixture of 7% (w/v) pullulan, 3% (w/v) linear polyacrylamide and 0.3 % (w/v) agarose was used as sieving matrix. Further experiments using only pullulan as a less complex matrix yielded a similar resolution with a much easier filling of the capillaries. | ||
| Fig. 6 Result of an SDS-CGE experiment performed with a voltage of 1000 V (duration 16 min). A mixture of 6 different fluorescent high molecular weight marker proteins (total amount 0.5 µg, Sigma) was separated in 15 % (w/v) pullulan and a running buffer of 0.1 M CHES, 0.1 M Tris, 0.1% (w/v) SDS. The picture is composed of a cascade of microscopic images taken by a fluorescent microscope combined with a CCD camera at different positions on the chip system. | ||
Fig. 6 was composed by a series of pictures taken by a fluorescent microscope (Olympus BX60) combined with a CCD camera (Kappa DX 30) at different positions on the chip. The laboratory setup of the instrument which is under development by Herolab allows a simpler automated detection of the fluorescence signals, however, with a slightly reduced sensitivity. Fig. 7 shows a result of a similar experiment (cf.Fig. 6) recorded by the optical system integrated in the instrument. A further advantage of the automated detection within the chip handling system is the time-resolved detection mode with a fixed detection line which can be placed at various positions depending on the desired separation length. The intensity diagram shown in Fig. 7 was obtained with the detection line located 10 mm away from the entrance to the separation channels. Positioning the detection line at the end of the capillaries, i.e. after a separation length of 60 mm, yields an increased resolution, however, accompanied by a loss of sensivity due to diffusional broadening of protein bands and a reduction of fluorescence intensity going along with that (data not shown).
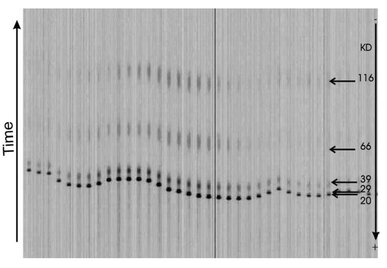 | ||
| Fig. 7 Result of a similar separation as described in Fig. 6 and detection with the optical system of the instrument. The detection line was located 10 mm away from the entrance to the separation channels. | ||
Fig. 8 shows a result of a CGE run of Alexa 488 labeled myoglobin obtained for a separation length of 60 mm. The result basically demonstrates the homogeneous electrophoretic conditions ranging over hundreds of capillaries, a fundamental prerequisite for highly parallel detection within the micro channels. Irregularities observed in first experiments are most likely attributed to air bubbles or dust in the nut or in parts of some capillaries. Thus, extreme care has to be taken during fabrication, chip preparation, e.g. gel filling, and chip handling.
 | ||
| Fig. 8 1.25 µg Alexa 488 labeled myoglobin from horse (4 ng per capillary) in a hot stacking gel solution (0.5% agarose in 0.5 M Tris-Cl pH 6.8) were filled into the connection nut between the first and the second dimension. As sieving matrix with a composition of 15% (w/v) pullulan and a running buffer of 0.1 M CHES, 0.1 M Tris-Cl pH 8.8, 0.1% (w/v) SDS was used. The experiment was performed in only 20 min at a maximum voltage of 2000 V. The detection was performed with an integration time of 800 ms and an interval time of 1601 ms per step. | ||
In summary, the results show the successful high-voltage separation of protein mixtures in polymer-based microfluidic channels in the presence of SDS. The time needed for separation of large proteins can be reduced using a lower pullulan concentration. Thus, a tailor-made separation matrix assures high resolution and good sensitivity for the particular target proteins under study. Furthermore, the integrated optics allows for a scanning of the whole chip. While this may result in scanning images with relatively low contrast it is of great use to monitor the separation and to optimize the separation length, i.e. to determine the optimum position of the detection line.
Transfer
The most challenging task in the development of a miniaturized 2D electrophoresis system is the sample transfer from the first to the second dimension, i.e. the transfer via an opening of 50 µm (shown in Fig. 1) from the IEF cavity into the 300 micro channels where the CGE is carried out. A high resolution in CGE requires a narrow, highly focused initial protein distribution. To that end, the protein transfer was simulated for different electrode configurations and switching modes. Fig. 9 shows the arrangement of the three electrodes needed for the protein transfer. A direct transfer (from electrode II to electrode III) results in a large tailing, and the microchannels become overloaded due to the high protein concentration in the focused IEF bands. The basic idea to overcome this problem is to transfer proteins into the micro channels and to redraw most of the transferred sample in a way that only a fraction is left within the channels. Fig. 10 shows a corresponding simulation result achieved by Lagrangian particle tracking. Assuming a negligible impact of the protein concentration on the electrical field strength, the field is computed for a fixed potential at each electrode. The particles represent ensembles of proteins having a common mobility and are driven according to the computed electrical field. The simulation data are used for identification of switching modes and estimation of switching times and voltages. Due to the simplified approach used, a quantitative correspondence to experimental results is not to be expected. However, as shown in Fig. 11, first experimental results prove the feasibility of the developed switching protocol. In particular, it is apparent that the molecules are collected in a comparatively narrow band and that the transfer process is approximately homogeneous over the different channels. In contrast to that, the results displayed in Figs 6 to 8, obtained with two electrodes only (II and III in Fig. 9), were based on much smaller initial amounts of protein sample. Further tests are currently under way.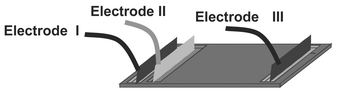 | ||
| Fig. 9 Arrangement of the electrodes during transfer and CGE. | ||
 | ||
| Fig. 10 Simulation of protein transfer. Each particle represents an ensemble of proteins. The arrow indicates the transport direction controlled by the applied voltages of the three electrodes (not shown) on top and at the right, left sides of the lower channel. | ||
 | ||
| Fig. 11 Seperation result of transferred Alexa 488 labeled myoglobin in 15% pullulan, 100 mM CHES, 100 mM Tris-Cl pH 8.8 and 0.1% SDS with an arrangement of three electrodes. The detection line was located 5 mm away from the IEF cavity (integration 800 ms per interval 1601 ms per line per 10 min). The transfer took place without the usage of a stacking gel. | ||
Summary and conclusions
We presented a miniaturized 2D-CGE device and discussed the basic steps towards a functional prototype, i.e. conceptual development, fabrication and experimental validation. For simplicity of handling and a later automation an integrated, i.e. non-modular set-up was chosen. The fabrication of the disposable device involves hot embossing of the micro-structured part, conventional micromachining and laser ablation of the main chip and solvent bonding for the connection of both parts. The complete fabrication process (etching of master structures into silicon, electroplating, polymer processing) was optimized in order to guarantee a high quality and reproducibility of the polymer parts.In the next steps both separation concepts, IEF and CGE, were tested separately. Both methods had to be adapted to the particular needs and conditions required for on-chip usage. In the context of IEF the IPG-strip support material was changed to a permeable polymer net in order to facilitate the protein transfer. Furthermore, an appropriate storage and rehydration cassette was developed. Among others, the implementation of CGE into the PMMA chip requires particular means in order to suppress EOF. Here, a specialized coating (Serva Electrophoresis GmbH, Heidelberg, Germany) turned out to be most suitable.
Most challenging in the development is the protein transfer from IEF to CGE separation. Based on computer simulated protein transfers, first experiments proved the general feasibility of the chosen approach. Thus, an important link in experimental validation could be closed. Future work will be dedicated to quantitative performance evaluations and further optimizations. On the one hand, the present design of the 2D chip and the handling system has a large flexibility concerning resolution, sensitivity and analysis times and thus can be easily optimized for certain applications. On the other hand, the system facilitates a highly automated protein analysis using disposable polymer chips suitable for mass production.
The presented system bears a large potential concerning a significant reduction in analysis time for 2D-electrophoresis and of an enhanced throughput. It is hoped that this will speed up protein analysis. Furthermore, the chip system bears the potential of subtractive analysis of patient samples for diagnostic purposes.
Acknowledgements
This work was supported by the BMBF grant “Analysis of the human proteome” (FKZ 01GG9836).References
- E. Verpoote, Electrophoresis, 2002, 23, 677–712 CrossRef CAS.
- R. Tonge, J. Shaw, B. Middleton, R. Rowlinson, S. Rayner, J. Young, F. Pognan, E. Hawkins, I. Currie and M. Davidson, Proteomics, 2001, 1, 377–396 CrossRef CAS.
- J. S. Fey and P. M. Larsen, Curr. Opin. Chem. Biol., 2001, 5(1), 26–33 CrossRef.
- X. Chen, H. Wu, C. Mao and G. M. Whitesides, Anal. Chem., 2002, 74(8), 1772–8 CrossRef CAS.
- F.-T. A. Chen, J. Chromatogr., 1991, 559, 445–454 CrossRef CAS.
- J. Wang, M. Pumera, M. P. Chatrathi, A. Escarpa, R. Konrad, A. Griebel, W. Dörner and H. Löwe, Electrophoresis, 2002, 23, 596–601 CrossRef CAS.
- J. Bhardwaj and H. Ashraf, Proceedings to SPIE: Micromachining and Microfabrication Process Technology, 1995, 224–233 Search PubMed.
- F. Lärmer and A. Schip, US Patent #5501893, 1996.
- H. Löwe, H. Mensinger and W. Ehrfeld, Jahrbuch Oberflächentechnik, Metall-VerlagHeidelberg, 1994, 77–95 Search PubMed.
- N. Grubhofer, German Patent #3032069, 1982.
- B. Radola, H. Schwall and M. Demharter, US Patent #5672416, 1989.
- A. Görg, W. Postel, R. Westermeier, E. Gianazza and P. G. Righetti, J. Biochem. Biophys. Methods, 1980, 3, 273–284 CrossRef CAS.
- P. G. Righetti, Immobilized pH gradients. Theory and methodology, Elsevier, Amsterdam, New York, Oxford, 1990 Search PubMed.
- A. Görg, W. Postel, S. Günther and J. Weser, in Electrophoresis '86, ed. J. M. Dunn, VCH, Weinheim, 1986, pp. 435–449 Search PubMed.
- S. Hu, L. Zhang, L. M. Cook and N. J. Dovichi, Electrophoresis, 2001, 22, 3677–3682 CrossRef CAS.
Footnotes |
| † Present address: Progen AG, Maaßstrasse 30, D-69123 Heidelberg, Germany. |
| ‡ Present address: IonGate Biosciences GmbH, Paul-Ehrlich-Str. 17, D-60596 Frankfurt, Germany. |
| § R. Konrad died in April 2001. |
| This journal is © The Royal Society of Chemistry 2004 |