Donor–acceptor polythiophene copolymers with tunable acceptor content for photoelectric conversion devices
Marinella
Catellani
*a,
Silvia
Luzzati
a,
Natalia-O.
Lupsac
a,
Raniero
Mendichi
a,
Roberto
Consonni
a,
Antonino
Famulari
b,
Stefano Valdo
Meille
b,
Francesco
Giacalone
c,
José L.
Segura
c and
Nazario
Martín
*c
aIstituto per lo Studio delle Macromolecole, CNR, Via Bassini 15, I-20133, Milano, Italy. E-mail: m.catellani@ismac.cnr.it
bDipartimento di Chimica, Materiali e Ingegneria Chimica, Politecnico via Mancinelli7, I-20131, Milano, Italy
cDepartamento de Química Orgánica, Universidad Complutense, Ciudad Universitaria, E-28040, Madrid, Spain
First published on 21st November 2003
Abstract
The synthesis and characterization of a new series of substituted polythiophenes containing an electron acceptor anthraquinone moiety in the side chain are reported. The acceptor molar content was varied by the co-polymerization of both alkylthiophene and thiophene bearing anthraquinone monomers in different ratios. NMR analysis shows a good correlation between the monomer feed composition at the beginning of the polymerization and the actual unit composition of the backbone. The conjugation length and the chemical composition of the copolymers as a function of the molecular weight have been studied by size exclusion chromatography. Small angle X-ray scattering and UV-Vis absorption spectra have been used to monitor the degree of order and chain organization in the solid state. The materials exhibit a lamellar organization, in which the anthraquinone units of neighboring side chains are also organized to some degree in layered structures parallel to the polythiophene main chains. The photoluminescence measurements in solution suggest that, upon photoexcitation of the polythiophene backbone, the anthraquinone moieties act as electron acceptors and the conjugated backbone as electron donor. The tunability of the donor–acceptor ratio and the morphology in the solid state make these photoactive copolymers interesting candidates for organic photoelectric conversion devices.
Introduction
Conjugated polymeric materials have been extensively studied in both academic and industrial laboratories due to their potential commercial applications in molecular-scale electronics. Some of the applications include light-emitting diodes,1 electrochromic displays,2 field-effect transistors,3 and integrated circuits.4 More recently polymeric materials received attention for the fabrication of low-cost and large area photoelectric conversion devices such as photodetectors or solar cells.5Photoelectric conversion in organic materials is based on the photoinduced electron transfer process involving two species with different ionization potential and electron affinity. The active layer of polymeric devices is a donor–acceptor bulk-heterojunction in which a conjugated donor polymer is mixed with acceptor molecules (i.e. fullerene) or macromolecules.6,7 In these photoactive composites the charge separation occurs at the donor–acceptor interface and the carriers are transported to the electrodes through the n- and p-type conducting networks formed by the two phase-segregated species: the positive charge via the polymeric domains, the electrons via the acceptor domains. The formation of an interpenetrating donor–acceptor phase is necessary for reaching the required device efficiency; for this reason the engineering of the blend morphology is quite important for the applications.8
An approach to enhance both the photoinduced charge transfer and the transport processes is the preparation of macromolecules in which an electron acceptor moiety is covalently linked to a hole transporting conjugated backbone.9 The so called “double cable” materials show larger donor–acceptor interfaces and are in principle ambipolar, having two different conduction pathways for electron and hole transport. The preparation of a p-type conjugated backbone carrying n-type conducting moieties allows the modulation of the donor–acceptor ratio and their interaction as a function of the macromolecule chemical structure. Furthermore, soluble and compatible ‘double cable’ polymers can also be obtained with a high content of acceptor, and can be easily processed in thin photoactive layers. Thiophene- and phenylenevinylene-based conjugated polymers bearing acceptor substituents such as fullerene10–12 or tetracyanoanthraquinodimethane13 have been recently described, and their potential use for photovoltaic devices has been investigated.14
With the aim to prepare processable photoactive materials with tunable properties we have prepared a new series of donor–acceptor polythiophene copolymers containing anthraquinone moieties as acceptor in the backbone side chains (see Fig. 1). We have designed these macromolecules and choose both donor polymer and acceptor species following these guidelines: i) polythiophene electronic properties can be easily tailored by functionalization of the monomers;15 ii) the anthraquinone molecule acts as an acceptor with a flat geometry helping to a 3-D assembling and then the formation of a pathway for the transport of electrons through the photoactive layer;16 iii) the copolymerization of two monomers is a powerful strategy for the chemical modification of the conjugated materials as well as for controlling their electronic properties.17 Polythiophenes containing anthraquinone as pendant functional groups have been previously prepared via electrochemistry18 or by chemical functionalization of a precursor polyalkylthiophene.19 These materials show the electrochemical reduction of the anthraquinone moiety and the n- and p-doping of the polythiophene backbone, thus they substantially retain the ground state properties of the individual thiophene backbone and anthraquinone moieties.
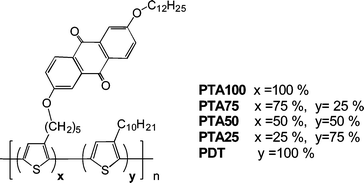 | ||
| Fig. 1 Chemical structure of donor–acceptor polythiophene copolymers containing electron accepting anthraquinone moiety. | ||
We have synthesized a series of regio-random polythiophenes by copolymerization of two monomers with FeCl3 in well defined ratios: a 3-decylthiophene and a thiophene ring bearing an anthraquinone moiety as substituent. The peculiar feature of these copolymers (PTAs) is that the content of acceptor can be easily tuned giving soluble and processable materials even with a 100% of anthraquinone substitution on the polymeric backbone.
The materials exhibit, in the solid state, spectral evidence of a photoinduced electron transfer process from the polythiophene backbone to the anthraquinone moieties as shown by FTIR photoinduced absorption and light-induced electron spin resonance experiments.20 The number of photogenerated charges and their lifetimes are easily tuned by tailoring the content of acceptor substituents in the polymer as well as the number of photogenerated charges and their lifetimes. In this paper we report the synthesis and macromolecular characterization as well as an absorption and emission study of these novel donor–acceptor copolymer series.
Experimental
3-Decylthiophene was synthesized following the method reported in the literature.21 Thiophene monomer 4 containing anthraquinone (see Scheme 1) was prepared in two steps from commercially available anthraflavic acid 1 by a Williamson etherification reaction with dodecyl bromide under stoichiometric control to yield 2 followed by reaction with thiophene derivative 322 in acetone by using potassium carbonate as base and sodium iodide as catalyst (yield 75%). 1H-NMR spectra of molecule 4 (CDCl3, 200 MHz): δ = 8.18 (d, 2 H, J = 7.6 Hz), 7.69 (d, 2 H, J = 1.2 Hz), 7.27 (s, 1 H), 7.24–7.21 (m, 2 H), 6.94 (d, 2 H, J = 4.5 Hz), 4.13 (t, 4 H, J = 6.4 Hz, -CH2-OCAr), 2.68 (t, 2 H, J = 6.8 Hz), 1.95–1.65 (m, 8 H, -CH2-), 1.60–1.26 (m, 18 H), 0.88 (t, 3 H, -CH3). 13C-NMR spectra (1,1,2,2-tetrachloroethane, 400 MHz): δ = 183.8, 165.7, 144.4, 137.3, 131.2, 129.9, 128.4, 126.9, 122.4, 121.5, 112.4, 111.1, 78.8, 77.6, 76.9, 69.5, 69.1, 31.9, 30.2, 30.0, 29.7, 29.6, 29.5, 29.4, 29.3, 29.2, 28.9, 28.8, 25.9, 25.6, 22.7, 14.0. FTIR (KBr). ν = 2924, 2852, 1664, 1637, 1490, 1396, 1305, 1238, 1157, 1122, 775 cm−1. Homopolymers and copolymers were prepared via chemical oxidation with FeCl3 in chloroform23 starting from mixtures of monomer 4 and 3-decylthiophene in a well defined molar ratio. We prepared homopolymer PTA100 endowed with an anthraquinone moiety for each thiophene ring (see Fig. 1), a poly-3-decylthiophene PDT and three copolymers, PTA75, PTA50 and PTA25, starting from monomer feeds containing 75%, 50% and 25% mol respectively of thiophene 4 bearing the acceptor moieties . The polymers prepared in this way (yields of about 90–95% by weight respect to the monomers) were washed several times with methanol and were extracted in a soxhlet apparatus with methanol in order to remove the residual oxidant. The purified homo- and copolymers are soluble in tetrahydrofuran, chloroform and carbon disulfide.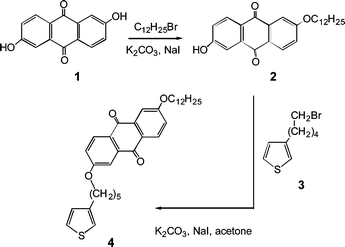 | ||
| Scheme 1 Synthesis of thiophene monomers containing an anthraquinone moiety. | ||
The NMR spectra were recorded at both room and high temperature, on Bruker Avance DRX and DMX spectrometers, operating at 9.39 T and 11.7 T respectively, both equipped with z-gradient probe. Proper relaxation times were used in order to obtain a quantitative analysis of the proton spectra. Gradient enhanced HMQC and HMBC hetero-correlated 2D experiments24 were acquired by using enhanced pulse sequences, with 100 ms as the long range evolution time. In order to obtain spectra at high temperature (350 K), the solvent used was deuterated 1,1,2,2-tetrachloroethane for all samples. Spectra were acquired, processed and plotted with XWIN-NMR Bruker software. 1H NMR spectra of copolymer PTA50: δ 8.09 (br s, CHAr), 7.58 (br s, CHAr), 7.11 (br, CHAr), 6.95 (br, CHTh), 4.05 (br, CH2), 2.73–2.54 (br m, CH2), 1.74–1.21 (br m, CH2), 0.81 ppm (br s, CH3); and 13C NMR selected data: δ 183.82 (CHAr), 165.67 (CHAr), 144.38 (CHTh), 137.34 (CHAr), 131.25 (CHAr), 129.94 (CHTh), 128.45 (CHAr), 126.9 (CHTh), 122.43 (CHAr), 121.66 (CHTh), 112.41 ppm (CHAr). Elemental analysis of copolymer PTA50: C 76.05%, H 7.16%, S 8.34%, O 8.45%.
The molecular weight distribution (MWD) and the on-line UV-Visible spectrum were obtained by means of a modular multi-detector size exclusion chromatography (SEC) system from Waters. The multi-detector SEC chromatography system consisted of an Alliance 2690 separation module (degasser, pump and injector), a column oven, a 410 differential refractometer (DRI) and an additional 996 UV-Visible diode array detector (DAD). The on-line DAD detector records the UV spectrum (wavelength from 190 to 800 nm, 1.2 nm of resolution) at each retention time after the SEC fractionation of the sample. The column set was composed of two PLGel Mixed C columns, 5 µm of particle size, from Polymer Laboratories (Shropshire, UK). The experimental conditions consisted of chloroform as mobile phase, 35 °C, 0.8 ml min−1 flow rate, 200 µl injection volume and approximately 0.25 mg ml−1 sample concentration. The SEC calibration curve, third order polynomial fit, was generated by means of eighteen narrow MMD polystyrene standards with the peak molecular weight Mp ranging from 162 to 3.2 × 106 g mol−1 obtained from the Polymer Standard Service.
Wide angle X-ray diffraction patterns of polymer powders pressed in a glass capillary were collected using graphite monochromated Cu-Kα radiation, (λ = 1.54179 Å), on a Bruker P4 diffractometer equipped with a HiStar 2D detector. Small angle X-ray scattering (SAXS) patterns of powders of the homopolymers and of copolymers all in glass capillaries, were collected on Bruker Nanostar system, equipped with mirror focusing and a HiStar 2D detector, with sample to detector distance of about 600 cm.
Electronic absorption spectra were performed with a Perkin Elmer Lambda 9 spectrophotometer on chloroform solutions or spin coated films on glass.
Photoluminescence (PL) spectra of polymeric solutions were recorded using 450 nm light excitation from a xenon lamp and a monochromator coupled to a N2 cooled CCD detector.
Results and discussion
NMR spectroscopy
In order to assign completely the 1H and 13C resonances of the copolymer, monomer 4 and homopolymer PTA100 have been used as starting models. Complete and unambiguous assignment has been obtained by means of 2D GE-HMBC and GE-HMQC experiments: the use of long range hetero-nuclear coupling constants were helpful in resolving spectral ambiguities.The determination of the proportions between electron-donor and electron-acceptor moieties in the materials is important to correlate the chemical composition with the photophysical properties of the materials. 1H NMR characterisation let us assess whether the average composition of the copolymers is in agreement with the composition of the starting monomer mixture.25 The ratio of 3-decylthiophene and monomer 4 units can be estimated by comparing the intensities of the protons in the α position to the thiophene rings (CH2 a in Scheme 2) at 2.8 ppm originating from both the monomer units, with the intensity of aliphatic protons closer to the anthraquinone moiety (CH2 b in Scheme 2) at 4.1 ppm originating only from monomer 4.
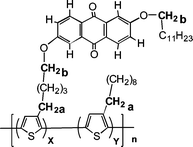 | ||
| Scheme 2 Chemical structure of PTA copolymers. | ||
In Fig. 2 the aliphatic and aromatic regions of copolymer PTA75 (upper) and homopolymer PTA100 (lower) are reported: the area of the proton resonances at 2.8 and 4.1 (indicated by the arrows) were used for quantitative evaluation of the copolymer composition. The anthraquinone content in mol% with respect to the thiophene rings of PTA75, PTA50 and PTA25 estimated by NMR were 81%, 53% and 28%. All the copolymers show a reasonable correlation between the composition of the monomer feed at the beginning of the polymerisation and the real unit composition of the backbone.
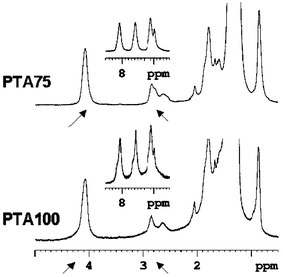 | ||
| Fig. 2 1H NMR aromatic and aliphatic regions of PTA75 (upper) and PTA100 (lower). The proton resonances at 2.8 and 4.1 ppm used for the evaluation of D–A content are marked by arrows. | ||
At room temperature both the homo- and copolymers were not completely dissolved in TCE and gave rise to aggregates, while at 350 K all the macromolecules were in solution. We have followed the modification induced by the temperature increasing from 300 to 350 K in the 1H NMR spectra and in the UV-Visible spectra of PTA100 homopolymer. Fig. 3 shows the evolution of the aromatic NMR signals at increasing temperature and the inset reports the electronic absorption spectra. By increasing the temperature from 300 K, the line width of the aromatic resonances becomes sharper with a small low field shift, thus reflecting the better solubility and the reduction of the aggregated forms present at room temperature. The electronic absorption spectra of PTA100 at room temperature (see inset in Fig. 3) shows a structured band due to the π–π* transition of the conjugated backbone with maxima at 490, 530, and 580 nm that indicate the presence of aggregated macromolecules.26 By heating the solution the structure of the band progressively disappears and the maximum shifts to the higher value of 450 nm indicating complete dissolution of the macromolecules.
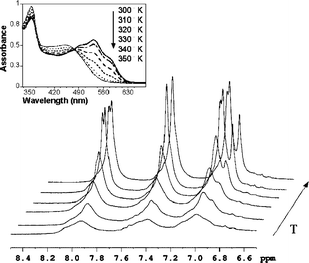 | ||
| Fig. 3 Evolution of 1H NMR aromatic signals of PTA100 during heating from 300 K to 350 K. In the inset are the electronic absorption spectra with increasing temperature. | ||
Size exclusion chromatography (SEC)
The molecular weight distribution, MWD, of the PTA100 homopolymer and of the three copolymers was estimated in a first approximation by a conventional SEC system. As usual the SEC system was calibrated using some narrow MWD polystyrene (PS) standards. It is well known that using such a SEC method the estimated MWD was not absolute but relative. Specifically, the relative calibration to PS standards presents two drawbacks. The first one derives from the difference in hydrodynamic volume between the PS polymer and the conjugated copolymers. In fact, it is well known that the SEC columns fractionate the macromolecules on the basis of the hydrodynamic volume and not the molecular weight. The second one derives from the possible non homogeneity in chemical composition of the copolymers. However, it is important to note that, although the MWDs obtained from the conventional SEC method were not absolute but relative, their comparison was meaningful. In addition, previous studies on similar polymers and/or copolymers have demonstrated that the difference between relative MWD and true MWD, obtained by absolute methods such as light scattering or mass spectrometry, were not very large.27 Consequently, we consider the recovered MWDs acceptable at least as a first approximation.Table 1 summarizes the most important results obtained by SEC: the molecular weight of the peak of the chromatogram Mp, the weight-average molecular weight Mw and the dispersity index D, and the wavelength of the electronic absorption maximum, λmax, obtained with the on-line DAD detector.
| Polymer | M p/kg mol−1 | Mw/kg mol−1 | D | λ max/nm |
|---|---|---|---|---|
| PTA100 | 39.8 | 45.4 | 5.7 | 440.0 |
| PTA75 | 54.5 | 83.6 | 4.2 | 443.6 |
| PTA50 | 62.6 | 160.1 | 5.7 | 442.4 |
| PTA25 | 94.1 | 221.8 | 5.5 | 442.2 |
Considering the very broad multi-components MWD of PTA100, in which coexist aggregates, polymer and several oligomers, the classical macromolecular parameters Mw and D are not particularly adequate for a comparison of the different samples. Probably, for this goal the Mp parameter, i.e. the molecular weight of the more abundant macromolecule population, is more meaningful. Considering that the molecular weight is relative to PS and not absolute, we could assert that Mp of the PTA100 homopolymer roughly corresponds to 70 repeating units. In an analogous way, starting from the Mp data reported in the Table 1 the number of repeating units of the three copolymers PTA75, PTA50 and PTA25 were approximately 114, 160 and 300 when the content of anthraquinone decreases in the macromolecule.
In SEC chromatography the use of an on-line diode array detector allows the UV-Visible spectrum to be collected in real time for each fraction of the eluting polymer. For polythiophenes this is a very powerful method to investigate the backbone conjugation length as a function of molecular weight and the homogeneity of the chemical composition of the copolymers.17,27,28
Fig. 4 shows the 3D plot of the PTA100 native homopolymer obtained by using the UV-Visible diode array detector. This plot contains a high information content regarding composition and conjugation of the backbone: the x-y-z coordinates are the retention time, the wavelength and the absorbance respectively, and it is possible to extract an electronic spectrum, from 190 to 800 nm with 1.2 nm of resolution, at each chromatographic retention time.
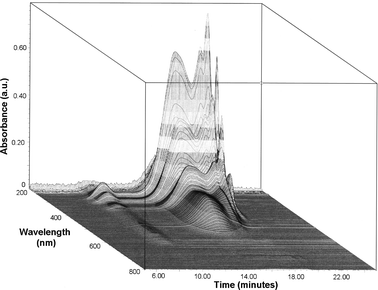 | ||
| Fig. 4 3D plot from the UV-Vis diode array detector of the PTA100 homopolymer. | ||
Fig. 5 shows the chromatogram, i.e. the refractometer signal, of the native PTA100 homopolymer. Considering only the polymeric peaks we can see in Fig. 5 three different components: the fraction of aggregate macromolecules marked with the label 1 at short retention times, the more abundant fraction of macromolecules marked with the label 2, and finally three oligomers marked progressively from 3 to 5. In Fig. 6 are reported the UV-Visible spectra of the different molecular weight fractions of PTA100 marked with the same progressive labels from 1 to 5 of the chromatogram in Fig. 5. The electronic spectra display a series of absorption bands in the UV region originating from the anthraquinone moieties,29 and a band in the visible region due to the π–π* transition of the polythiophene whose maximum depends from the mean conjugation length of the backbone.
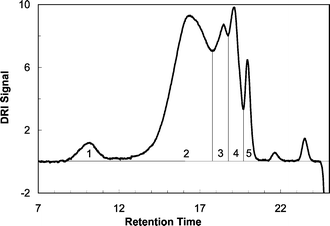 | ||
| Fig. 5 Chromatogram, refractometer signal, of the PTA100 homopolymer. A label (progressively from 1 to 5) denotes various fractions with different molecular weight. | ||
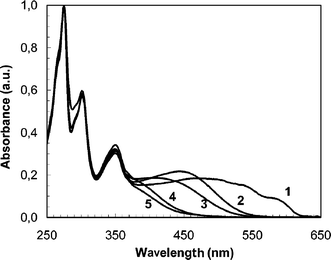 | ||
| Fig. 6 UV-Visible spectra of the different chromatogram fractions of the PTA100 obtained with a diode array detector. The spectra are marked with the same labels as the fractions of Fig. 5. | ||
Fraction 1 shows a structured spectrum with absorption maxima, λmax, at 500, 540 and 590 nm that correspond to aggregated macromolecules typically present in polythiophenes also in a good solvent such as chloroform.17,27,28 Fraction 2 shows a maximum at 450 nm typical for polyalkylthiophene solutions; comparing the spectra of each retention time in this chromatogram interval no meaningful variation of the absorption features was observed. This means that there is a relatively good homogeneity of the chain chemical composition in this more abundant fraction of PTA100. The spectra of the oligomeric fractions 3, 4 and 5 show a blue shift of the π–π* band due to the lowering of the conjugation length as expected for low molecular weights. Such analysis of the SEC measurement driven for the homopolymer could be extended also to the three copolymers although the comparison of the UV-Visible spectrum of their various components is not reported here.
Using the on-line diode array detector a qualitative analysis of the aggregates typically present in solution for these polymers can be made. Fig. 7 shows the chromatograms of the copolymers obtained with the intensity of the UV-Visible signal at λ = 254 nm (the absorption of the thiophene rings). Macromolecule aggregates are clearly present as a shoulder in the chromatogram of PTA100 (see interval labeled 1 in Fig. 5) and of PTA75, the copolymer with higher molar content of anthraquinone. On the contrary, the presence of aggregates in the PTA25 and PTA50 copolymers is not evident either in their chromatograms or absorption spectra. From a qualitative point of view the extent of aggregation in PTA75 is high as in the homopolymer, and strongly decreases in the copolymer with the lower anthraquinone content.
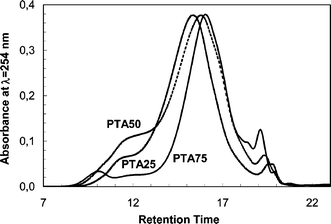 | ||
| Fig. 7 Comparison of the UV-Vis signal, λ = 254 nm, of the homopolymer and of three copolymers. | ||
X-Ray diffraction
Small angle X-ray scattering (SAXS) patterns, recorded from PTA100 samples and from the copolymers are shown in Fig. 8.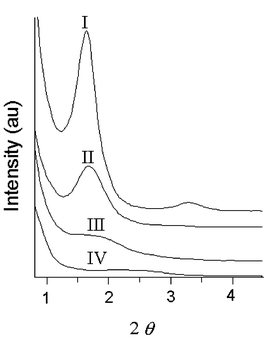 | ||
| Fig. 8 SAXS data for PTA100 (curve I), PTA75 (curve II), PTA50 (curve III), PTA25 (curve IV). | ||
For the homopolymer a well defined, sharp maximum is apparent at 2θ ≈ 1.68° corresponding to ca. 53 Å. A second order is apparent at 2θ ≈ 3.3° (26.5 Å). SAXS patterns of PTA75 show a much weaker maximum, shifted to a slightly lower periodicity, of 50 Å and a shoulder at about 26 Å. The pattern of copolymers PTA50 shows a weak peak at about 48 Å while SAXS of PTA25 shows very weak and broad maxima centered around 2θ ≈ 2.4°.
The 53 Å periodicity shown by the homopolymer is consistent with a lamellar organization strongly reminiscent of the structures of form I poly-3-alkylthiophenes: in fact this value can be extrapolated for linear alkyl side chains containing 29–30 carbons from literature values of the form I a lattice parameter of polyalkylthiophenes30 (see Fig. 13 of reference 30). It is notable that the side chains of PTA100 contain 30 non-hydrogen atoms and the volume requirements can be reasonably expected to be comparable or slightly smaller than for polyalkylthiophenes. The fact that also a clear second order is apparent is consistent with a remarkable degree of long range order. From the peak width at half maximum we can estimate lamellar stacks of ca. 6 molecular layers in the case of the homopolymer.
The reduction of the spacing to 50 Å in the case of PTA75 could be consistent with the inclusion of a limited number of copolymeric units with lower volume requirements (i.e. 3-decylthiophenes) in the lamellar domains. On the other hand the lower relative intensity of the SAXS maximum in the case of PTA75 as compared to the homopolymer indicates a lower degree of lamellar organization in the copolymer. An average stack size of ca. 250 Å suggests in this case aggregates reduced to an average of 5 layers.
An essential contribution to establish reasonable structural and morphological models comes from the wide angle diffraction (WAXD) data presented in Fig. 9. PTA100 powder patterns show at high angle only a broad amorphous maximum, indicating negligible crystallinity and very limited degrees of organization at length scales of a few angstroms. This is consistent with the DSC analysis, which does not show any glass transition or melting point for the polymer. The very large contribution to the total scattering of the low angle maxima for PTA100 in Fig. 9 suggests on the other hand that the long range organization of the material at the scale of 3–20 nm is very high: it could be viewed as a layered mesophase.
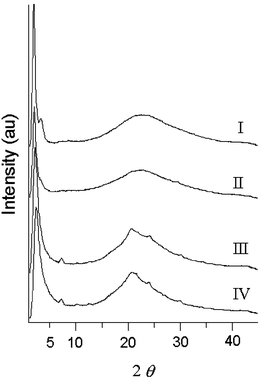 | ||
| Fig. 9 WAXD data for PTA100 (curve I), PTA75 (curve II). PTA50 (curve III), PTA25 (curve IV). | ||
Very weak maxima can be evidenced at about 12–13 Å and 10.5 Å. Similar observations apply to the PTA75 pattern although the intensity contribution of the small angle maximum at ca. 50 Å to the total scattering intensity is much lower confirming a much less extensive lamellar organization. The WAXD patterns of PTA50 and PTA25 present relatively broad maxima at low angle implying lamellar organization respectively with 49 and 37 Å periodicities. However the patterns surprisingly reveal, especially for PTA25, non-negligible crystallinity due to the poly-3-decylthiophene (PDT) component. Both PTA25 and PTA50 present weak but clear maxima at ca. 12.4, at 8.7 and 7.1 Å representing respectively the third, forth and fifth order of a 35–36 Å lamellar spacing. We therefore think that the 49 Å value found for PTA50 is very likely to arise from the contribution of unresolved maxima corresponding to ca. 52 Å and ca. 35 Å. The broadening of the low angle maximum in the case of PTA50 could be explained in part by these considerations. Whether a similar explanation also applies to PTA25 and for the lowering of the spacing from 53 to 50 Å in the case of PTA75 is doubtful. In the latter case there is hardly evidence of crystalline PDT and in the former case self organization of the anthraquinone substituted component is unclear. Consistent with the plausible inclusion of a limited number of the longer side chains in the lamellar structures and non-regioregularity, the lamellar spacing for PTA25 is substantially higher than the one reported for form I of regioregular poly(3-decylthiopene).
The evidence for phase separation of the two components implies a significantly blocky nature for the copolymers we are discussing. On the other hand the diffraction data evidence that the systems are not simply a mixture of the pure homopolymer phases: thus their copolymeric nature is supported. Electron microscopy investigations on the system are in progress and will be reported elsewhere.
We are unable at this stage, to give quantitative estimates of the volume fraction of the homopolymer PTA100 organized in lamellar stacks: it is likely to be high although we can expect variations depending upon the film deposition conditions, which may influence also the size of the stacks and the degree of short range order within the lamellar structure. Considering the implications of lamellar organization, it seems quite probable that the anthraquinone units of neighboring sidechains will organize to some degree in layered structures parallel to the polythiophene main chain layers. The weakness of significant features in the wide angle pattern of PTA100 indicates clearly that the degree of local ordering must remain low.
Electronic absorption
The UV-Visible absorption spectra of the PTA series in chloroform solution and in films are reported in Fig. 10. The spectra display an absorption band in the visible region which is due to the π–π* transition of the polythiophene backbone, and a series of bands in the UV region, originating from the anthraquinone moieties.31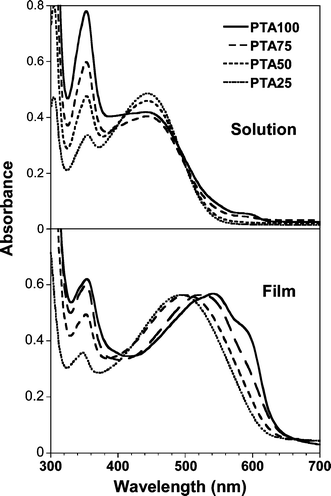 | ||
| Fig. 10 Electronic absorption spectra of the PTA series in chloroform solution with 3.31 × 10−4 M concentration (upper) and in spin coated films on glass (lower). | ||
In the spectra of the solutions the intensities of both polythiophene and anthraquinone bands (upper of Fig. 10) depend on the chemical composition of the macromolecules. PTA100 chloroform solution shows an absorption maximum at 450 nm, that is 10–20 nm higher than the usual value of regiorandom polyalkylthiophene in a good solvent, and for the copolymers λmax were close to 440 nm. This feature can be due to the presence of macromolecule aggregates, which is evident only in the spectra of PTA100 and PTA75, or to a more planar conformation of the conjugated backbone in solution with respect to polyalkylthiophene prepared with FeCl3.
The features of the solid state electronic spectra are also affected by the copolymer chemical composition (bottom of Fig. 10). A red shift of the backbone absorption maximum is observed upon increasing the anthraquinone content, suggesting that the substituents are inducing a better backbone conjugation and/or a better three-dimensional organization of the copolymer chains.
The homopolymer PTA100 displays the most red shifted spectrum, with a λmax at 540 nm. This feature can be reasonably ascribed to a better order due to the homogeneity of the side chains, as confirmed by X-ray analysis. However, considering that the λmax of PDT films, with the same degree of regioregularity of the homopolymer, is at 510 nm, we infer that in PTA100 the long side chains incorporating the anthraquinone residues play a specific role in driving the chain ordering.
Photoluminescence
We have studied the photoluminescence properties after photoexcitation at 450 nm of our donor–acceptor materials in chloroform solution with a dilution adjusted to have the same number of absorbed photons at the excitation wavelength. All the copolymer spectra show an emission maximum at 576 nm. Fig. 11 shows a comparison between the photoluminescence (PL) intensity of the PTA series with mixtures of PDT and PTA100 in the same molar ratios of the copolymers. The PL of donor–acceptor copolymer is quenched by 1–2 orders of magnitude with respect to PDT and the quenching increases with the anthraquinone content. This suggests that electron transfer from the photoexcited polythiophene backbone to anthraquinone is occurring and that the charge transfer is fast enough to compete with the radiative recombination of the excitons. This indicates that when the hole conducting backbone and the acceptor molecules are covalently linked they are closer and can interact better, giving a more efficient photoinduced electron transfer process. It is interesting to note that in this case an energy transfer from the polymer to the acceptor cannot explain the PL quenching because the electronic transitions of the anthraquinone are at higher energies with respect to the π–π* transition of the conjugated backbone. This is a relevant difference respect to solutions of conjugated polymers and fullerene, where the energy transfer from the chain to the acceptor has been identified as an important step of the photoexcitation mechanism of these D–A systems.32 Moreover we have found less efficient PL quenching for the mixtures of electron donor PDT and PTA100 than in the copolymers.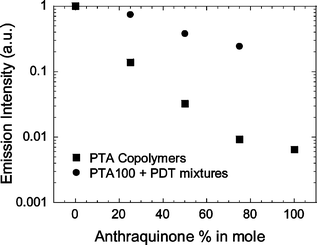 | ||
| Fig. 11 Comparison between the photoluminescence intensity of PTA samples in chloroform solution, and mixtures of PDT and PTA100. PL intensity is normalized to the number of absorbed. | ||
Conclusions
We have synthesized a series of regiorandom polythiophenes, PTA, bearing an anthraquinone moiety electron acceptor in the side chains. The copolymerisation of 3-decylthiophene units and thiophene containing anthraquinone monomers in a well defined ratio permits a modulation of the chemical composition of the macromolecule. In these materials the content of acceptor can be easily tuned giving soluble and processable materials even with a 100% of anthraquinone substitution on the backbone units.We have measured the amount of the electron acceptor in the polymer series by NMR spectroscopy and we found a good correlation between the composition of the monomer feed at the beginning of the polymerisation and the effective unit composition of the backbone.
The molecular weight distribution was measured by size exclusion chromatography, and the backbone conjugation length as a function of molecular weight was studied for all the polymers. SEC analysis using an on-line diode array detector has evidenced that the chemical composition for each copolymer is independent from the molecular mass.
Small angle X-ray scattering pattern of PTA100 is consistent with an extensive lamellar organization similar to the structures of form I of polyalkylthiophenes, but the ordering at a local scale (∼0.5 nm) is quite low. Considering the lamellar organization of the chains the anthraquinone units of neighboring sidechains are bound to also organize in layered structures parallel to the polythiophene main chains, which in their own turn will show some degree of stacking. Above concentrations around 50% of the alkylthiophene units in the copolymers, crystallization of increasing fractions of the material in a polydecylthiophene lattice is evidenced, suggesting a blocky nature of the copolymers.
Electronic absorption spectra of the PTA series show that the π–π* transition band of the conjugated backbone is slightly red shifted with respect to a regiorandom polyalkylthiophene both in solution and in the solid state. Moreover a red shift of the chain absorption maximum is observed upon increasing the anthraquinone content, thus suggesting that the substituents are inducing a better backbone conjugation and/or a better three-dimensional organization of the copolymer chains. The PTA100 electronic absorption feature and the SAXS measurement strongly suggest that the anthraquinone moieties are better organized in the solid state. This organization of the acceptor part of the macromolecule is promising for the formation of n-type pathway for the electron transport in the solid state.
The PL of donor–acceptor copolymer in solution is quenched by 1–2 orders of magnitude with respect to polydecylthiophene and the quenching increases with the anthraquinone content. This suggests that electron transfer from the photoexcited polythiophene backbone to anthraquinone is occurring. Moreover, it is possible to tune the photoinduced charge transfer by changing the donor–acceptor ratio with the chemical composition of the chain.
In conclusion, these materials with tunable donor–acceptor content, are soluble and processable, exhibit the photoinduced electron transfer process in solution and an organization of acceptor moieties in the solid state that promote the formation of a pathway for the electron transport. These macromolecules are attractive for organic photoelectric conversion device applications, their study in photodiodes is in progress and will help in clarifying their potentiality as photoactive materials.
Acknowledgements
This work was supported by the European Commission (EUROMAP RTN project nr. HPRN-CT-2000-00127), the MCYT of Spain (Project BQU2002-00855) and by the Comunidad de Madrid (project 07N/0004/2002).References
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burn and A. B. Holmes, Nature, 1990, 347, 539 CrossRef CAS.
- P. Schottland, K. Zong, C. L. Gaupp, B. C. Thompson, C. A. Thomas, I. Giurgiu, R. Hickman, K. A. Abboud and J. R. Reynolds, Macromolecules, 2000, 33, 7051 CrossRef CAS.
- H. Sirringhaus, N. Tessler and R. H. Friend, Science, 1998, 280, 1741 CrossRef CAS.
- B. Crone, A. Dobalapur, Y.-Y. Lin, R. W. Filas, Z. Bao, A. LaDuca, R. Sarpeshkar and H. Katz, Nature, 2000, 403, 521 CrossRef CAS.
- (a) C. J. Brabec, N. S. Sariciftci and J. C. Hummelen, Adv. Funct. Mater., 2001, 11, 15 CrossRef CAS; (b) J. Nelson, Curr. Opin. Solid State Mater. Sci., 2002, 6, 87 CrossRef CAS.
- G. Yu, Y. Gao, J. C. Hummelen and A. J. Heeger, Science, 1995, 270, 1789 CAS.
- J. J. M. Hall, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498 CrossRef CAS.
- S. E. Shaheen, J. C. Brabec, F. Padinger, T. Fromherz, J. C. Hummelen and N. S. Sariciftci, Appl. Phys. Lett., 2001, 78, 841 CrossRef CAS.
- T. Benincori, E. Brenna, F. Sannicolò, L. Trimarco, G. Zotti and P. Sozzani, Angew. Chem., Int. Ed. Engl., 1996, 35, 648 CrossRef CAS.
- A. M. Ramos, M. T. Rispens, J. K. J. van Duren, J. C. Hummelen and R. A. J. Janssen, J. Am. Chem. Soc., 2001, 123, 6714 CrossRef CAS.
- A. Cravino, G. Zerza, H. Neugebauer, M. Maggini, S. Bucella, E. Menna, M. Svensson, M. R. Andersson, C. J. Brabec and N. S. Sariciftci, J. Phys. Chem. A, 2002, 106, 70 CrossRef CAS.
- F. Zhang, M. Svensson, M. R. Andersson, M. Maggini, S. Bucella, E. Menna and O. Inganas, Adv. Mater., 2001, 13, 1871 CrossRef CAS.
- (a) G. Zerza, A. Cravino, H. Neugebauer, N. S. Sariciftci, R. Gómez, J. L. Segura, N. Martín, M. Svensson and M. R. Andersson, J. Phys. Chem. A, 2001, 105, 4172 CrossRef CAS; (b) F. Giacalone, J. L. Segura, N. Martín, N. M. Catellani, S. Luzzati and N. Lupsac, Org. Lett., 2003, 5, 1669 CrossRef CAS.
- A. Cravino and N. S. Sariciftci, J. Mater. Chem., 2002, 12, 1931 RSC.
- J. Roncali, Chem. Rev., 1997, 97, 173 CrossRef CAS.
- T. Yamamoto and H. Etori, Macromolecules, 1965, 28, 3371.
- M. Catellani, P. C. Stein, S. Luzzati, R. Mendichi and A. Giacometti Schieroni, Polymer, 1996, 37, 1059 CrossRef CAS.
- J. A. Crayston, A. Iraqi, P. Mallon, J. C. Walton and D. P. Tunstall, Synth. Met., 1993, 55-57, 867 CAS.
- A. Iraqi, J. A. Crayston, P. Mallon and J. C. Walton, J. Mater. Chem., 1998, 8, 31 RSC.
- S. Luzzati, M. Scharber, M. Catellani, N. Lupsac, F. Giacalone, J. L. Segura, N. Martín, H. Neugebauer and N. S. Sariciftci, Synth. Met., 2003, 139, 731 CrossRef CAS.
- K. Tamao, S. Komada, I. Nakajima and M. Kumada, Tetrahedron, 1982, 38, 3347 CrossRef CAS.
- P. Bäuerle, F. Wurthner and S. Heid, Angew. Chem., Int. Ed. Engl., 1990, 29, 419 CrossRef.
- R. Sagimoto, S. Takeda, H. B. Gu and K. Yoshino, Chem. Express, 1986, 1, 635 Search PubMed.
- W. Wilker, D. Liebfritz, R. Kerssebaum and W. Bermel, Magn. Reson. Chem., 1993, 31, 287.
- G. Lanzani, A. Piaggi, C. Botta, M. Catellani, E. Prevosti and P. C. Stein, Solid State Commun., 1996, 99, 707 CrossRef CAS.
- S. D. D. V. Rughooputh, S. Hotta, A. J. Heeger and S. F. Wudl, J. Polym. Sci., Part B: Polym. Phys., 1987, 25, 1071 CAS.
- R. Mendichi, A. Bolognesi, Z. Geng and A. Giacometti Schieroni, Proceedings of International GPC Symposium ‘94, Orlando, FL, USA, 1994, pp. 827–838 Search PubMed.
- A. Bolognesi, F. Bertini, R. Consonni, R. Mendichi, A. Giacometti Schieroni and A. Provasoli, Acta Polym., 1997, 48, 507 CrossRef CAS.
- A. Bolognesi, R. Mendichi, A. Giacometti Schieroni and D. Villa, Macromol. Chem. Phys., 1997, 198, 3277 CrossRef CAS.
- S. V. Meille, V. Romita, T. Caronna, A. J. Lovinger, M. Catellani and L. Belobrzeckaja, Macromolecules, 1997, 30, 7898 CrossRef CAS.
- M. Büschel, C. Stadler, C. Lambert, M. Beck and J. Daub, J. Electroanal. Chem., 2000, 24, 484.
- P. A. van Hal, J. Knol, B. M. W. J. Langeveld-Voss, S. C. J. Meskers, J. C. Hummelen and R. A. J. Janssen, J. Phys. Chem. A, 2000, 104, 5974 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2004 |