Highly sensitive two-photon chromophores applied to three-dimensional lithographic microfabrication: design, synthesis and characterization towards two-photon absorption cross section
Youmei
Lu
*a,
Fuyuki
Hasegawa
a,
Takamichi
Goto
b,
Satoshi
Ohkuma
b,
Setsuko
Fukuhara
b,
Yukie
Kawazu
a,
Kenro
Totani
a,
Takashi
Yamashita
b and
Toshiyuki
Watanabe
a
aDivision of Applied Chemistry (Graduate School), 2-24-16 Naka-cho, Koganei-city Tokyo, 184-8588, Japan. Tel: +81-42-388-7289
bDepartment of Pure and Applied Chemistry, Faculty of Science and Technology, Tokyo University of Science, 2641 Yamazaki, Noda, 278-8510, Chiba, Japan. E-mail: luym19@cc.tuat.ac.jp; toshi@cc.utat.ac.jp; Tel: +81-47-124-9067
First published on 14th October 2003
Abstract
A series of D–π–A–π–D type chromophores were synthesized by the dehydration reaction of 4-R2N-benzaldehye (R = Ph, Bun, Et, Me) and diaminomaleonitrile (corresponding to the chromophores 1, 2, 3 and 4, respectively), in which a polar imino double bond (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) replaced the double bond (–CHCH–) in the π-conjugated centers. Femtosecond laser induced fluorescence intensity was used to evaluate two-photon absorption (TPA) cross sections, δ, using a USB-2000 CCD. Results show a change of terminal groups from Ph2N– to Me2N– influenced the δ value significantly through a change of the quantum yield, φ. However, the two-photon absorption peak position was only slightly affected. The chromophores 2 and 3 were found to afford polymers in the presence of the functional triacrylate monomer at low laser power at 755 and 820 nm. This demonstrated that the enhanced δ value was not a main factor in the improvement of chromophore two-photon photosensitivity. Such information can be useful in the design of more efficient two-photon chromophores for imaging and power-limiting applications.
N–) replaced the double bond (–CHCH–) in the π-conjugated centers. Femtosecond laser induced fluorescence intensity was used to evaluate two-photon absorption (TPA) cross sections, δ, using a USB-2000 CCD. Results show a change of terminal groups from Ph2N– to Me2N– influenced the δ value significantly through a change of the quantum yield, φ. However, the two-photon absorption peak position was only slightly affected. The chromophores 2 and 3 were found to afford polymers in the presence of the functional triacrylate monomer at low laser power at 755 and 820 nm. This demonstrated that the enhanced δ value was not a main factor in the improvement of chromophore two-photon photosensitivity. Such information can be useful in the design of more efficient two-photon chromophores for imaging and power-limiting applications.
Introduction
Two-photon absorption is attracting considerable attention owing to its various applications in the areas of three-dimensional imaging, optical data storage and three-dimensional lithographic microfabrication (3DLM) through two-photon-induced polymerization (TPIP).1–7 This is due to the fact that simultaneous two-photon absorption requires a very high photon flux, which is only present at the point of the focus. Thus the TPI polymerization is confined to the focal volume. This high spatial resolution contributes to the ability of TPIP not only to scan the laser in the x and y direction but also to change the focal plane (z) without overwriting existing features. Therefore, 3DLM is obtained by a single processing step. 3D polymeric structures include a photonic band gap structure, waveguide structures and a micro-channel structure.8–11 Recently, microrotator and microtweezers have been manufactured by TPIP of less than 0.4 µm and 0.25 µm in diameter, respectively.12,13 Efficient two-photon absorbers based on bis(styryl)benzene constructs bearing electron-donating moieties were synthesized by Albota14 and Bunning15 and their co-workers, and found to undergo a presumed two-photon induced electron transfer to highly functional free-radical polymerizable acrylate monomer. On the other hand, Belfield and Pitts conducted two-photon-induced polymerization under near-IR fs-laser irradiation by using a commercially available UV photoinitiator, consisting of the rather involved syntheses of bis(dialkylamino)stilbene family.16–21 In this case, the acrylamine, acrylate and acrylic acid system was induced to polymerize in the presence of N,N-dimethylaniline, triethanolamine or an onium salt as the co-initiator wherein the co-initiator amine acted as the electron donor upon chromophore excitation. However, the photosensitivity was relatively low due to the low two-photon absorption of the UV initiator.The molecules considered in this work have the general structure D–π–A–π–D, wherein D is a terminal group, π is a phenylimine function and A is an electron-accepting cyano group. Since cyano groups are used as the side groups, the increase of electron transfer leads to a significant increase in the δ value relative to electron-donating methoxy groups (as in compound 5).14 Additionally, the change of the conjugated backbone through the introduction of the polar imino groups will allow the tuning of the linear and nonlinear optical responses of quadrupolar derivatives as a result of the influence of the chromophoric two-photon absorption properties.22,23 Both polar groups (–CHN–) will enhance the extent of the charge transfer from the terminal groups to the center, correlated with the two cyano-groups. Upon chromophoric excitation, the π-conjugated center acts as an electron acceptor, and intramolecular energy and electron transfer will occur more readily from the terminal R2N– groups than via intermolecular energy and electron transfer.16–21 Radical forms can be easily generated in the terminal groups. Moreover, the solubility of the chromophore improves with the replacement of –CHCH– by the polar flexible –CHN– group. Fig. 1 shows the molecular structures 1–4 studied in this paper. Due to the fact that they can be synthesized in one-step with a high yield and initiate polymerization of triacrylate monomer at a high level of sensitivity, makes application in two-photon-induced polymerization possible.
 | ||
| Fig. 1 The structures of the molecules studied in this work. Compounds 0 and 5 were reported by Perry's group.10,14,22 | ||
Experimental
1. Chemicals
Diaminomaleonitrile (98%) and 4-diphenyl- or 4-dialkyl-aminobenzaldehydes (98%) were purchased from Aldrich. The solvents used in the fluorescence studies were spectrophotometric grade and were checked by fluorescence spectroscopy to make sure they contained no fluorescent impurities.2. Instruments
Absorption spectra were measured with JASCO V-560 UV/Vis spectrophotometer, accurate to ±2 nm. Luminescence spectra were obtained with JASCO FP-6500/6600 for windows spectrofluorimeter. The measurements of δ values were calibrated relative to Rhodamine-B (abbreviated as R-B) with absolute TPA cross-section determined by Xu and Webb in the 690–1050 nm range.24The samples were dissolved in toluene and chloroform at a concentration of 5 × 10−5 M to reduce the effect of reabsorption, and δ values were determined through the two-photon-induced fluorescence method using a one-arm setup (Ti-Saphire laser: 85-fs pulses at a repetition rate of 82 MHz (Spectra-Physics Tsunami)). The laser beam was focused over the lens (NA = 0.3) at a laser power of 1.0–2.5 mW and the fluorescence was collected at right angles with respect to the incident beam. One used method was the conventional method where the fluorescence was collected by a lens and passed through a monochromator and measured by a photomultiplier tube (PMT) as described by Rumi et al. (in toluene).22 The second used method was measuring a spectrograph with a CCD (in CHCl3). In the latter case, the diameter of the focused laser beam is ca. 3.2 µm, and the diameter of the sensor of the CCD is ca. 50 µm. The fluorescent light in the cell underfilled the sensitive area of the detector. Thus, the fluorescence can be measured even though the emission is weak. The measurements were carried out in a laser intensity range for which a quadratic dependence of the fluorescence signal on the laser was observed. Moreover, to reduce the path length of the fluorescence emission in the solution under study and affect the reabsorption, the incident beam was directed as close as possible to the windows of the cell on the side where the light was collected.
Cyclic voltammetry was performed under argon with tetrahydrofuran solutions ca. 10−4 M in sample and 0.1 M in [Bun4N]+[PF6]−, using a glassy carbon working electrode, a platinum auxiliary electrode, and a silver–silver ion electrode which was easily constructed by putting a silver wire in a solution of 0.01 M AgClO4 in tetrahydrofuran. Potentials were referenced by the addition of ferrocene to the cell. Fluorescence lifetime measurements were carried out by the single-photon counting method. The sample was dissolved in CH3CN excited at 337 nm by using a nitrogen laser (Nihon laser LN-100 pulse width of 300 ps).
3. Synthesis
The synthetic route is shown in Fig. 1. The chromophores were prepared from the dehydration reaction of 4-diphenyl- or 4-dialkyl-aminobenzaldehyde and diaminomaleonitrile. The synthetic procedures for obtaining the chromophores 1–4 were the same as used in the work by Yu et al.25 All quadrupolar molecules were characterized by nuclear magnetic resonance (1H NMR), elemental analysis (Yanako MT-5 model) and liquid chromatography mass spectrometry (SHIMADZU), which confirmed the assigned structures.Results and discussion
1 Core effects
The role of π-conjugation on the δmax value can be seen in Table 1. For the chromophore 1, the change of π-conjugation, –CHN– replacing –CHCH–, led to δmax decreasing five-fold with the quantum yield φ reduced to half the value relative to compound 0,14 while its UV absorption peak red-shifted around 70 nm. This is indicative of improved electronic conjugation in 1. However the experimental results showed that the increase in the molecular polarity is unfavorable to the enhancement of two-photon absorption and the quantum yield as seen from Table 1.
2 Solvent effects
A summary of optical data and parameters of chromophores 1–4 in toluene and CHCl3 are listed in Tables 2 and 3. Since chromophore 4 did not dissolve in toluene, its two-photon properties were not investigated in that solvent.| Chromphore | 1 λ ex/nm | 2 λ ex/nm | δ/GM | φ | φ δ |
|---|---|---|---|---|---|
| 1 | 542 | 810 | 510 | 0.34 | 173 |
| 835 | 495 | ||||
| 2 | 541 | 800 | 395 | ||
| 830 | 430 | 0.06 | 26 | ||
| 3 | 537 | 800 | 330 | ||
| 820 | 340 | 0.03 | 12 |
| Chromphore | 1 λ ex/nm | 2 λ ex/nm | δ/GM | φ | φ δ |
|---|---|---|---|---|---|
| 1 | 552 | 840 | 180 | ||
| 885 | 265 | 0.46 | 120 | ||
| 2 | 553 | 820 | 490 | 0.1 | 49 |
| 840 | 480 | ||||
| 3 | 547 | 820 | 545 | 0.05 | 29 |
| 840 | 520 | ||||
| 4 | 536 | 820 | 2050 | 0.02 | 31 |
| 840 | 1910 |
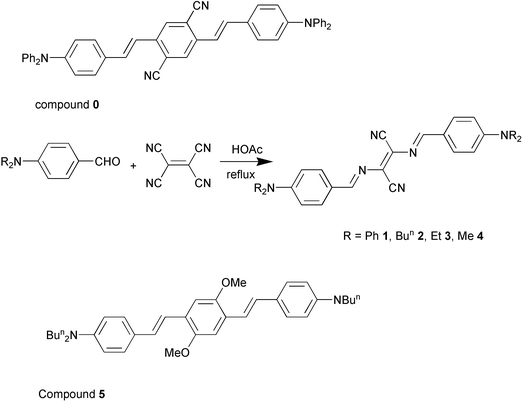
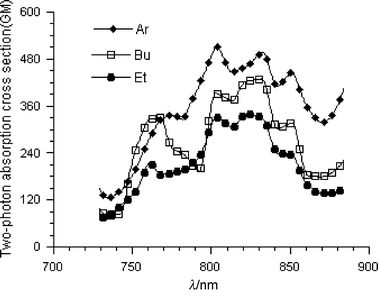
For chromophore 1, with Ph2N– as the terminal group, the δ value was enhanced significantly in the apolar solvent, toluene, rather than the polar solvent, CHCl3. By contrast, the experimental results show that the δ values of chromophores 2 and 3 were large in CHCl3 relative to those in toluene as seen in Tables 2 and 3 and Figs. 2 and 3. In the case of the chromophore 1, four terminal phenyl groups decrease the molecular polarity through the extended π-conjugation. As a result, two-photon absorption and excited fluorescence was enhanced in toluene even in the presence of the strong electron-accepting groups –CHN– and –CN. The enhancement of two-photon absorption of the chromophores 2 and 3 in the polar solvent was attributed to the increase of the molecular polarity from the –CHN– and –CN groups in the center. It was thus concluded that the two-photon proprieties were dependent upon the solvent polarity in a significant way.
 | ||
| Fig. 2 Two-photon-induced fluorescence excitation spectra of chromophores 1–3 in toluene at a concentration 5 × 10−5 M. | ||
 | ||
| Fig. 3 Two-photon-induced fluorescence excitation spectra of chromophores 1–4 in CHCl3 at a concentration 5 × 10−5 M. | ||
3 Terminal groups effects
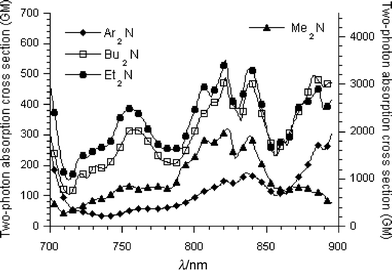
As listed in Table 3, the change of the terminal group from Ph2N– to Me2N– produced different effects on the δ value. The positions of 1λex were slightly influenced as shown in Fig. 4. Based on the experimental results that the φδ value was similar from Ph2N to Me2N–, the significant difference in the δ value is mainly attributed to the change of the quantum yield. That is to say, the terminal groups modified the TPA through the effects on φ. In particular, the Ph2N– terminal groups led to a red-shift of the two photoabsorption peaks, relative to Bu2N–, Et2N– and Me2N– with an obvious reduction in two-photon absorption and an increase in the quantum yield. This is mainly attributed to the increase in the extent of electronic delocalization by attaching four phenyl rings on both ends. As for chromophore 4, its large δ value was due to its relatively small quantum value. | ||
| Fig. 4 UV/Vis absorption spectrum recorded for chromophores 1–4 in CHCl3. | ||
4 Microfabrication and sensitivity by TPIP
To demonstrate the utility of these two-photon chromophores, we attempted to prepare two-photon cross-linkable semi-solid acrylate resins, to include the chromophores 1–5 at molar concentration of 5 × 10−4 M. The resins consisted, by weight, of 70% multifunctional acrylate monomer (SR9008![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶SR368 = 1∶1, purchased from Sartomer Co. Ltd.) and 29.9% poly(styrene-co-acrylonitrile) as a polymer binder, as described in the literature.26 The resin mixture was dissolved in dioxane, cast on to a glass substrate at a thickness controlled by a Baker Film Applicator, and allowed to set by slow evaporation of the solvent. The thickness of the cured films could be varied from 100 to 200 µm. In microfabrication experiments the laser beam was passed through a microscope (Olympus, IX50/IX70), and the samples were exposed by irradiation with tightly focused laser pulses (numerical aperture, NA = 1.2, ∼0.39 µm radial spot size at 755 nm and ∼0.45 µm at 820 nm) from a Ti:sapphire laser. The exposures were patterned under computer-control by shuttering the beam and translating the film relative to the focal point using a three-axis micropoistioner (Sutter, MP-285). Following exposure, the film was immersed in dimethylformamide to dissolve the non-irradiated resin leaving behind the cross-linked polymeric structure.
∶SR368 = 1∶1, purchased from Sartomer Co. Ltd.) and 29.9% poly(styrene-co-acrylonitrile) as a polymer binder, as described in the literature.26 The resin mixture was dissolved in dioxane, cast on to a glass substrate at a thickness controlled by a Baker Film Applicator, and allowed to set by slow evaporation of the solvent. The thickness of the cured films could be varied from 100 to 200 µm. In microfabrication experiments the laser beam was passed through a microscope (Olympus, IX50/IX70), and the samples were exposed by irradiation with tightly focused laser pulses (numerical aperture, NA = 1.2, ∼0.39 µm radial spot size at 755 nm and ∼0.45 µm at 820 nm) from a Ti:sapphire laser. The exposures were patterned under computer-control by shuttering the beam and translating the film relative to the focal point using a three-axis micropoistioner (Sutter, MP-285). Following exposure, the film was immersed in dimethylformamide to dissolve the non-irradiated resin leaving behind the cross-linked polymeric structure.
Chromophore 4 was not easily dissolved in the resin. As for chromophore 1, it readily dissolved in to the solution of the resin, but the dye was partly crystallized after the film dried. Therefore, the concentration of chromophores 1 or 4 in the resin was low relative to that of chromophores 2 and 3. As a result their two-photo polymerization photosensitivity was not investigated. There may be the similar reason why the bis(styryl)benzene chromophore 0 with bisphenylamino terminal groups with a large δ value was not used as a two-photon initiator as widely as that of 5 with bis-n-butylamino groups.14
Considering the chromophores 2 and 3 have a significant absorption peak at 755 nm (Fig. 3), the microfabrications shown in Fig. 5(a) and (b) and Fig. 6(a) and (b) were obtained at 755 and 820 nm, respectively. Here, we define the two-photon polymerization rates (Rp) based on the equation π(d/2)2vs, where d is the width of the written protruding line and the scanning speed vs is 50 µm s−1. The results are listed in Table 4. It was found that the sensitivity of the chromophores 2 and 3 increased at a peak of two-photon absorption, 820 nm, relative to that at 755 nm. Though the laser power at 755 nm (5.0 mW) is higher than that at 820 nm (3.0 mW), a higher efficiency of the polymerization occurred at 820 nm (cf. Fig. 5(b), 6(b) to Fig. 5(a), 6(a), respectively). However, at the same laser wavelength and power, the ratio of Rp of the chromophores 2 and 3 was not consistent with the ratio of δ, as listed in Table 4.
 | ||
| Fig. 5 Optical micrograph of the grating with 20 µm spacing using chromophore 2. (a) Excitation wavelength: 755 nm; laser power: 5.0 mW with 1.8 µm width; (b) excitation wavelength: 820 nm; laser power: 3.0 mW with 2.4 µm width. | ||
 | ||
| Fig. 6 Optical micrograph of the grating with 20 µm spacing using chromophore 3. (a) Excitation wavelength: 755 nm; laser power: 5.0 mW with 2.0 µm width; (b) Excitation wavelength: 820 nm; laser power: 3.0 mW with 3.4 µm width. | ||
| Chromophore | E D+/D/V | ΔG/eV | τ/ns | 2 λ ex | δ/GM | φ | d/µm | R p/µm3 s−1 |
|---|---|---|---|---|---|---|---|---|
| 2 | 0.05 | 0.58 | 0.3 | 755 | 315 | 0.1 | 1.8 | 120 |
| 820 | 490 | 2.4 | 230 | |||||
| 3 | 0.18 | 0.71 | 0.3 | 755 | 390 | 0.05 | 2 | 160 |
| 820 | 545 | 3.4 | 450 | |||||
| 5 | −0.01 | −0.14 | 1 | 730 | 900 | 0.88 | 3 | 395 |
The analysis of the oxidation potential for chromophores 2 and 3 (0.050 and 0.18 V, respectively) showed that Bu2N– is a strong donor relative to Et2N– in the ground state. This indicates that the highest occupied molecular orbital (HOMO) of the chromophore 2 is located at a higher energy than that of the chromophore 3. The free energy, ΔG, of the electron transfer reaction was evaluated from the relation ΔG = ED+/D − EAcc/Acc− − Eex, as cited in the literature,26 where ED+/D is the electrode potential of the chromophore 2 or 3, the electrode potential of the monomer acrylate, EAcc/Acc−, is −2.77 V, and Eex is the energy difference between the ground state and the lowest-lying excited state (S1) of the chromophore 2 or 3. Both the free energy values have been found to be positive as listed in Table 4.
In our case, the TPIP sensitivities of the chromophores 2 and 3 were demonstrated through increasing the scanning speed at an identical laser power and wavelength. Because of changes of laser power resulting from a slight change of the focus position in the z-axis direction it is difficult to investigate the threshold power at which the weakly polymerized resins become set following developing. As shown in Fig. 7(a) and (b), both microstructures were observed after development at a scanning speed of 290 µm s−1. However, comparing Fig. 7(a) and (b), we found the strength of the fabricated structures initiated by chromophore 3 is stronger than that by chromophore 2 at 290 µm s−1.
 | ||
| Fig. 7 Optical micrographs with 10 µm spacing: (a) using chromophore 2 with 2.4 µm width; (b) using the chromophore 3 with 3.4 µm width. Excitation wavelength: 820 nm; laser power: 3.0 mW. The scan speed was increased from 50 to 290 µm s−1 with a step of 30 µm s−1 from the left. | ||
In addition, Fig. 8 shows the microstructure obtained using compound 5 at a laser power of 3.0 mW, excitation wavelength 730 nm and with a scanning speed at 50 µm s−1. Taking into account a slight difference of the radial spot size of the focused beam at the different excitation wavelengths, the Rp value of the compound 5 is similar to that of the chromophore 3 at the identical laser power, 3.0 mW, as listed in Table 4.
 | ||
| Fig. 8 Optical micrograph of the grating with 20 µm spacing using compound 5. Excitation wavelength: 730 nm; laser power: 3.0 mW with 3.0 µm width. | ||
Compound 5 has been demonstrated to be a very efficient two-photon initiator among the bis(dialkylamino)stilbene family synthesized by Perry' group. For compound 5, it was reported that the bimolecular quenching rate with SR9008 was large (up to 3.7 × 108 M−1 s−1 along with a negative free energy, ΔG). Even though the experimental results showed that bimolecular quenching with SR9008 was not observed for chromophores 2 and 3, the high two-photon sensitivity of chromophores 2 and 3 means that electron-transfer occurred easily between the initiator and the monomer despite their positive ΔG and ED+/D relative to the compound 5. Provided that TPIP is a process of the free radical polymerization, the rate of gerenation of excited states is mainly influenced by TPA cross section, but the radical-generation quantum yield may be related to the quantum yield and the fluorescence lifetime of the chromophore. Otherwise, we observed that the chromophore 3, with the lowest quantum yield and the shortest fluorescence lifetime, can still initiate acrylate to polymerize with the highest sensitivity among the chromophores 2, 3 and compound 5. This indicates that they may be a differing main factor influencing the TPIP sensitivity rather than the reaction free energy value and the excited energy level.
Conclusion
For the series of chromophores studied here, the presence of strong electron acceptors in the center of π-conjugation led to a significant decrease in the values of φ and δ, compared to bis(dialkylamino)stilbene. It was demonstrated that the improvement of two-photon photosensitivity was not mainly consistent with high two-photon absorption efficiency. Based on the fact that chromophores 2 and 3 can be synthesized in one-step and can induce polymerization of acrylate for a wide range of laser wavelength (755 and 820 nm) suggests the practicality of broader use in 3DLM technology. Based on the fact that the chromophore 3 has a high sensitivity of TPIP, similar to that of compound 5, suggests it may replace compound 5, which requires a rather involved synthesis. In the development of 3DLM technology, in order to optimize the sensitivity of the two-photon initiators and monomer, such information may be useful in searching for chromophores with ideal properties for energy transfer and electron transport for imaging and power-limiting applications.Acknowledgements
We are grateful to Mrs Y. Takizawa and N. Okamura for the NMR measurements, and Dr H. Yamazawa and Dr T. Hasimoto for their discussion of LC-MS. This research was also supported by the Ministry of Education, Science, Sports and Culture, Japan, in order to fulfill the Grant-in-Aid for Scientific Research on Priority Areas (Molecular synchronization), 11167220, 2001–2003, Grant-in-Aid for Scientific Research (B), 14350128, 2003 and the 21st Century COE (Center of Excellence) program on “Future Nano-Material” (conducted at Tokyo University of Agriculture & Technology).References
- W. Denk, J. H. Strickler and W. W. Webb, Science, 1990, 248, 73 CAS.
- J. H. Strickler and W. W. Webb, Opt. Lett., 1991, 16, 1780 CAS.
- D. A. Parthenopoulos and P. M. Rentzepis, Science, 1989, 245, 843 CAS.
- J. H. Strickler and W. W. Webb, Proc. SPIE, 1990, 1398, 107 Search PubMed.
- E. S. Wu, J. H. Strickler, W. R. Harrell and W. W. Webb, Proc. SPIE, 1992, 1674, 776 Search PubMed.
- S. Maruo, O. Nakamura and S. Kawata, Opt. Lett., 1997, 22, 132 CAS.
- S. Maruo and S. Kawata, J. MEMS, 1998, 7, 411 Search PubMed.
- H.-B. Sun, V. Mizeikis, Y. Xu, S. Matsuo and H. Misawa, Appl. Phys. Lett., 2001, 79, 1 CrossRef CAS.
- M. P. Joshi, H. E. Pudavar, J. Swiatkiewicz, P. N. Pradad and B. A. Reianhard, Appl. Phys. Lett., 1999, 74, 170 CrossRef CAS.
- B. H. Cumpston, S. P. Ananthavel and J. W. Perry, Nature, 1999, 398, 51–54 CrossRef CAS.
- W. Zhou, S. M. Kuebler, K. L. Braun, J. W. Perry and S. R. Marder, Science (Washington, DC), 2002, 296, 1106 Search PubMed.
- S. Yokoyama, T. Nakahama, H. Miki and S. Mashiko, Appl. Phys. Lett., 2003, 82, 3221–3223 CrossRef CAS.
- S. Maruo, K. Ikuta and H. Korogi, Appl. Phys. Lett., 2003, 82, 133–135 CrossRef CAS.
- M. Albota, D. Beljonne and J. W. Perry, Science, 1998, 281, 1653–57 CrossRef CAS.
- T. J. Bunning, S. M. Kirkpatrick and D. W. Tomlin, Chem. Mater., 2000, 12, 2842–2844 CrossRef CAS.
- L. luo, C. Li and S. Feng, J. Opt. A: Pure Appl. Opt., 2001, 3, 489–492 Search PubMed.
- K. D. Belfield, J. liu and S. J. Andrasik, Polym. Prepr., 2001, 42(1), 713 Search PubMed.
- L. L. Brott, R. R. Naik and M. O. Stone, Polym. Prepr., 2001, 42(1), 675 Search PubMed.
- J. D. Pitts, P. J. Campagnola and S. L. Goodman, Macromolecules, 2000, 33, 1514–23 CrossRef CAS.
- P. J. Campagnola, D. M. Delguideice and S. L. Goodman, Macromolecules, 2000, 33, 1511–1513 CrossRef CAS.
- K. D. Belfield, X. Ren and E. J. Miesak, J. Am. Chem. Soc., 2000, 122, 1217–1218 CrossRef CAS.
- M. Rumi, J. E. Ehrliich and J. W. Perry, J. Am. Chem. Soc., 2000, 122, 9501–10 CrossRef CAS.
- B. A. Reinhardt, S. J. Clarson and P. N. Prasad, Chem. Mater., 1998, 10, 1863–1874 CrossRef CAS.
- C. Xu and W. W. Webb, J. Opt. Soc. Am. B, 1996, 13, 481–491 CAS.
- J. Yu, Z. Chen and T. Watanabe, Jpn. J. Appl. Phys., 2001, 40, 3201 CrossRef CAS.
- S. M. Kuebler, M. Rumi and J. W. Perry, J. Photopolym. Sci. Technol., 2001, 14, 656–668 Search PubMed.
| This journal is © The Royal Society of Chemistry 2004 |