Synthesis of tetrathiafulvalenes suitable for self-assembly applications†
Emma Dahlstedta, Jonas Hellberg*a, Rodrigo M. Petoral, Jr.b and Kajsa Uvdalb
aDept. of Chemistry, Organic Chemistry, Royal Institute of Technology, SE-100 44, Stockholm, Sweden. E-mail: jhel@kth.se; Fax: +46-8-791 2333; Tel: +46-8-790 8127
bLaboratory of Applied Physics, Department of Physics and Measurement Technology, Linköping University, SE-581 83, Linköping, Sweden
First published on 13th November 2003
Abstract
A series of new tetrathiafulvalenes, with double alkylthiol or alkyldisulfide substitution, have been prepared with a synthetic procedure that allows variation of different substituents. The target compounds 6a–e and 15e–i are sparsely soluble in organic solvents, but TTFs 6d and 15g gave a relatively dense packed monolayer upon exposure to gold surfaces.
Introduction
Tetrathiafulvalenes (TTFs) (1) are the most successful class of heterocycles in terms of creating highly conducting1 and superconducting organic crystalline materials.
Good intermolecular overlap between TTF π-stacks gives high charge carrier mobilities, especially when sulfur substituents can create a two-dimensional network of contacts, thereby suppressing Peierls-distortion.2 Many systems have been studied in detail and valuable insights in the solid state physics of organic materials have been achieved.3 These valuable properties have rarely been possible to transfer to more tractable systems, as in the case of oligothiophenes.4 Langmuir–Blodgett films of tetrathiafulvalenes have been studied and thin films have been prepared with interesting results,5 but few electronic devices such as field-effect transistors have been realized.
The self-assembly methodology, where alkylthiols self-assembles on an active metal surface, offers an alternative way of preparing ordered thin films of organic materials.6
If successful this would then open up the possibility of transferring the unique properties of tetrathiafulvalene charge transfer salts to thin film devices.7
Self-assembly monolayer (SAM) systems with tetrathiafulvalenes have previously been studied by several groups. TTF-SAMs were first reported by Yip and Ward8 but these systems were not particularly electrochemically stable. Bryce and co-workers studied a TTF with one alkylthiol attached to TTF substituted with a crown ether i.e. (2),9 which proved to yield more stable SAMs. A structural variation of 2 has been presented by Sallé and co-workers (3),10 as well as by Eschegoyen and co-workers (4).11
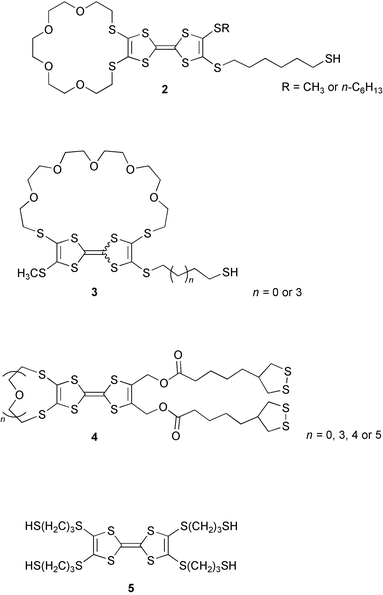
All three variations have been evaluated as electrochemical probes for different anions. Fujihara et al. have demonstrated that multiple short chain alkylthiol substituents can create highly stable SAMs (5).12
Other applications proposed for SAMs are for instance in tunneling devices.13
In order to be able to create a SAM that could work as an organic thin film field-effect transistor (OTF-FET) with appreciable charge carrier mobility, we should design a system that is able to form a stable SAM where a defined vertical region (layer) consists of a relatively densely packed electroactive π-system, e.g. TTF (Fig. 1).
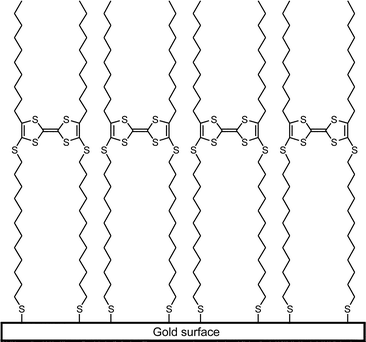 | ||
| Fig. 1 Attempted idealised TTF-SAM structure. | ||
The distances, both between the π-system and the surface, as well as “above” the electroactive region, should be possible to vary by the chosen synthesis. These regions should consist of alkyl- (sp3) chains, in order to isolate the conducting layer from the surface.
We envisaged that a tetrathiafulvalene system that could form a stable SAM should have at least two alkylthiol “legs” as well as considerable alkyl substitution “above” the TTF-layer. It was also found desirable to avoid statistic phosphite couplings in the synthesis of unsymmetrical tetrathiafulvalenes.
We therefore decided to synthesize tetrathiafulvalenes of type 6, which we anticipated to show several advantages but also a few drawbacks:
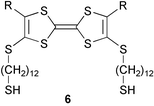
(i) The synthesis of 6 should allow relatively straightforward strategies for satisfying the demand for easy variation of the two different alkyl chains, as well as being a robust and reliable synthesis scheme.
(ii) The double alkyl thiols will render SAM stability, probably enhanced by a long upper alkyl chain, which also may self-assemble and form a stabilizing layer “above” the TTF π-stacking layer.
(iii) The two lower sulfur substituents should facilitate a more extended overlap between adjacent tetrathiafulvalenes, leading to higher charge carrier mobility.
(iv) The alkyl thiol chains are rather separated by the TTF-units, which can create a void in the SAM. This “space” perhaps has to be filled with additional free alkylthiols to develop a more uniform layer but in turn leading to more complicated SAM-studies.
The SAM-candidates of type 6 exist in equilibrium between a cis and trans diastereomer in the presence of acids.14 This can also make the kinetics of monolayer formation more difficult to study, but it might be possible to overcome this by performing the self-assembly under equilibrium conditions (low pH and elevated temperatures).
We present here the synthesis and full characterization of the self-assembly candidates, as well as some preliminary results on film formation.
Results and discussion
To create structures that would self-assemble, our first consideration was to choose a rather long alkylthiol chain that would attach to the substrate and preferably form an electronically insulating layer between the TTF units and the surface. In turn we could vary the R-group in 6 freely, with different alkyl chain lengths, to see if we could achieve more densely packed structures.The synthetic sequence, which is described in Scheme 1, is a modification of a previously published procedure.15
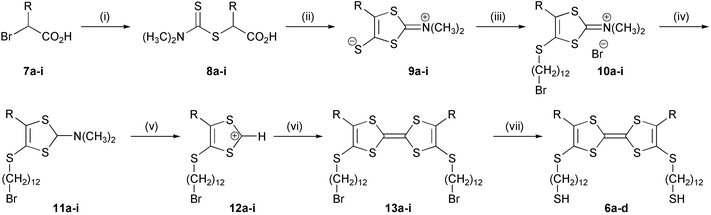 | ||
| Scheme 1 a: R = CH3, b: R = C2H5, c: R = C3H7, d: R = C4H9, e: R = C6H13, f: R = C10H21, g: R = C12H25, h: R = 1-naphthyl, i: R = 2-naphthyl. Reagents and conditions: (i) Me2NCS2Na, EtOH, 0 °C; (ii) Ac2O, Et3N, CS2, rt, acetone; (iii) Br(CH2)12Br, acetone, reflux; (iv) NaBH4, EtOH, rt; (v) HPF6, H2SO4, 0 °C; (vi) Et3N, CH3CN, rt, N2; (vii) (Me3Si)2S, (n-Bu)4NF, THF, −10° C. | ||
The α-brominated carboxylic acid underwent a substitution reaction when treated with the sodium salt of N,N-dimethyldithiocarbamic acid, to give 8a–i in 60–93% yield. The crystallinity varies with the chain length of the R-substituent. Compounds 8a–i were, without further purification, dissolved in acetone and treated with acetic anhydride and triethylamine. The presumed intermediate is a 4-oxyl mesoion, which underwent a [1,3]-dipolar cycloaddition with simultaneous release of carbonyl sulfide when carbon disulfide was added to the solution. The products 9a–i were obtained as yellow mesoionic precipitates in 40–96% yield. These products are stable at room temperature and can be stored at ambient temperature and atmosphere for several years without significant decomposition. Alkylation of 9a–i at the thiolate functionality was carried out in acetone with 1,12-dibromododecane in excess. The reaction proceeded smoothly at reflux in less than 6 h to yield 4-dodecylthio-1,3-dithiolium salts 10a–i in 72–99% yield. Reduction of 10a–i with sodium borohydride in ethanol16 to the corresponding amines 11a–i (61–91% yield) followed by deamination by treatment with concentrated sulfuric acid and hexafluorophosphoric acid, yielded 1,3-dithiolium hexafluorophosphates 12a–i. These compounds are highly hygroscopic and decompose quickly in air and were therefore reacted without further purification with triethylamine in dry acetonitrile, which gave TTFs 13a–i in fair yields after purification. On treating 13 with hexamethyldisilathiane and (n-Bu)4NF, according to a recent procedure,17 target compounds 6a–e were obtained. Due to the low solubility of these sticky products, they could not be purified by column chromatography and analyses of these compounds were not trivial. The structures were not suitable for NMR-characterization due to the low solubility in most organic solvents. The issue was further complicated by FAB-MS and MALDI-TOF spectra of products 6b–e, since they showed predominantly the (M − 2)+ ion (Fig. 2 shows the FAB-spectrum of 6b), which would suggest the presence of a disulfide (inter- or intramolecular) or other insaturations. However, treatment of 6b with DTT (dithiothreitol) which would have reduced all disulfide groups, left the mass spectrum unaffected.
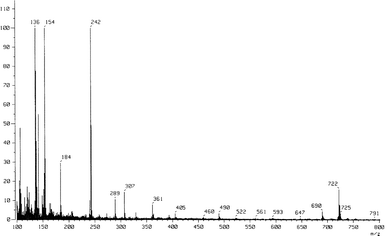 | ||
| Fig. 2 FAB-MS of 6b. | ||
The IR spectra showed a weak absorption at 2522 cm−1, indicating the presence of an S–H bond. Our speculation of the FAB-MS behavior is that the compounds interact (self-assembles) with a metal surface in the mass spectrometer, which can cause an oxidation to the dehydrogenated form. Moreover, elemental analysis confirmed that bromine had been substituted with sulfur. For TTFs 13e–i we used another approach to achieve our “target” molecules.18 Compounds 13e–i were converted to the corresponding disulfides by the reaction with a nucleophilic sulfur transfer agent 1419 according to Scheme 2, yielding target TTFs 15e–i as cyclic disulfides. The products, which precipitated from DMF, were filtered off and washed with diethyl ether to give products with a sticky apperance.
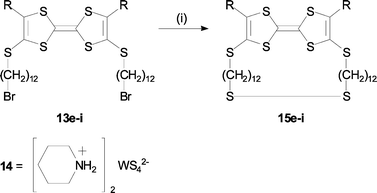 | ||
| Scheme 2 Reagents and conditions: (i) 14, DMF, 70 °C. | ||
The electrochemical behavior of TTFs 13a–i has been investigated by cyclic voltammetry (CV) in dichloromethane and the data are collected in Table 1.
| E1/V | E2/V | ΔE = (E2 − E1)/V | |
|---|---|---|---|
| a 1 mM (n-Bu)4NClO4 (0.1 M) in CH2Cl2, scan rate 500 mV s−1, E vs. SCE. | |||
| 13a | 0.43 | 0.78 | 0.36 |
| 13b | 0.42 | 0.78 | 0.36 |
| 13c | 0.41 | 0.77 | 0.36 |
| 13d | 0.42 | 0.78 | 0.36 |
| 13e | 0.42 | 0.77 | 0.36 |
| 13f | 0.42 | 0.78 | 0.36 |
| 13g | 0.42 | 0.77 | 0.36 |
| 13h | 0.49 | 0.78 | 0.29 |
| 13i | 0.50 | 0.81 | 0.30 |
Due to the low solubility of TTFs 6a–d and 15e–i in dichloromethane at room temperature no cyclic voltammograms could be taken from solution but this will be further investigated together with film forming properties. All measured cyclic voltammograms for compounds 13 exhibit two reversible one electron transfer processes corresponding to the successive formation of the cation radical (E1) and dication (E2) as expected for TTFs. The 1-naphthyl-substituted TTF 13h even showed a third oxidation as illustrated in Fig. 3. This may be due to oxidation of the naphthalene ring system, no corresponding reduction was however observed, indicating a more reactive, unstable oxidized species. As was expected, the values of the oxidation potentials and the difference ΔE = E2 − E1 were comparable with previously reported results for TTF (1), E11/2 = 0.33 and E21/2 = 0.71.20 What could be noted was that the naphthyl-substituted TTFs showed a slight positive shift of E1, compared to the alkyl-substituted TTFs, but similar potential of E2, that gave a 60–70 mV decrease of ΔE.
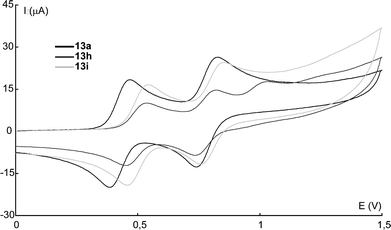 | ||
| Fig. 3 Cyclic voltammograms of TTFs 13a, 13h and 13i. | ||
The film forming ability of TTF-thiols 6d and 15g were tested. The n-butyl-substituted TTF 6d were immersed in toluene solution for a month, at room temperature and 70 °C, in the absence and presence of methanesulfonic acid. The corresponding dodecyl derivative 15g were also immersed for one month. The results from ellipsometry and advancing contact angles are shown in Table 2. Both TTFs gave monolayers of good and reproducible quality. It is clear from Table 2 that longer times and more acidic conditions do not alter the thickness of the film to any substantial degree. It is also clear that the film thickness is less than would be anticipated from a stretched molecular arrangement as in Fig. 1. Interestingly enough the longer alkyl chain on TTF 15g, does not result in a higher monolayer thickness.
| TTF Sample | D/Å | θa/° | θr/° |
|---|---|---|---|
| a In the presence of 0.5 μmol of (10 mol%) MeSO3H.b Incubated for 4 weeks (3 weeks at 70 °C + 1 week at rt).c Incubated for 1 week at rt.d Incubated for 48 h at 70 °C.e Incubated for 24 h at 70 °C. | |||
| 6db | 19.6 ± 0.3 | 75 | 63 |
| 6da,b | 19.2 ± 0.2 | 69 | 55 |
| 15gb | 15.0 ± 0.3 | 76 | 67 |
| 6dc | 16.0 ± 0.4 | 77 | 65 |
| 6dd | 15.9 ± 0.4 | 74 | 49 |
| 6da,e | 18.0 ± 0.3 | 63 | 53 |
The advancing contact angle of water on both type of monolayers indicates a more hydrophobic than hydrophilic surface due to the alkyl chains present on the topmost part of the organic film. These values do not indicate a high surface hydrophobicity, suggesting that the ring structure may somehow be on top of the film and partially influences the hydrophilicity of the surface. The advancing contact angle value is comparable to that of the HS(CH2)11OCH3 monolayer (θa = 74°) when adsorbed on gold.21 For comparison, the advancing contact angle value of HS(CH2)16CH3 is known to be 115°.21 The high hysteresis value (difference between advancing and receding contact angle) also indicates a more heterogenous outermost layer at the micron scale.
The thickness and contact angle measurement results suggest a film formation of 6d and 15g incubated for a month having densely packed alkylthiol chains on the surface, and with ring structures and alkyl tails exposed on the topmost part of the layer. The surface region is not well ordered, with the alkyl chains to some degree penetrating down into the alkylthiol region. A more definitive description and characterization of these monolayers will be possible after IR and XPS-studies which are now in progress.
Conclusions
The synthesis of a new series of tetrathiafulvalenes with either double alkylthiol (6a–e) or alkyldisulfide (15e–i) substitution has been accomplished. A versatile synthetic procedure is presented which allows a variety of substituents to be present. TTFs 6d and 15g gave relatively dense packed monolayers on gold, with inner alkyl chains more organized and outer alkyl-chains randomly organized with gauche defects.Experimental
Electrochemical measurments
Cyclic voltammetry was carried out at ambient temperature in CH2Cl2 at 500 mV s−1 with (n-Bu)4NClO4 (0.1 M) as electrolyte and measured vs. a SCE reference electrode. The half-wave potential for the ferrocene–ferrocenium couple was found to be 0.49 V in our system.Sample preparation for self-assembly
The substrate used is a 2000 Å thick electron beam evaporated-polycrystalline gold layer on Si(100) with 20–25 Å thick Ti as adhesion promoter. Prior to incubation, the gold samples were carefully cleaned to remove organic contamination. The substrates were incubated in the following toluene-based solutions (200 µM): 6d, 6d–10 mol% MeSO3H and 15g. The immersion periods were: 24 h, 48 h, 1 week and 1 month. The samples were kept in the dark and at either room temperature or heated at 70 °C during the incubation period. Ultrasonic rinsing with toluene for 5 min and N2 blow-drying of samples were carried out directly before mounting in measurement apparatus.Ellipsometry
Single-wavelength ellipsometry was performed using an automatic Rudolph Research AutoEL ellipsometer with He–Ne laser light source, λ = 632.8 nm, at an angle of incidence of 70°. The freshly cleaned gold sample substrates were measured prior to their incubation, and the collected average value of the refractive index was later used in a model “ambient/organic film/gold”, assuming an isotropic, transparent organic layer with a refractive index of n = 1.5. The film thickness is calculated as an average of measurements at five different spots on the sample of each compound.Contact angle goniometry
Contact angles were measured with Ramé-Hart NRL 100 goniometer, in air, i.e. without control on the humidity, using fresh MilliQ water. At least five measurements of advancing and receding contact angle were done per sample.Syntheses
NMR spectra were recorded on a Bruker AM 400 (400 MHz) or Bruker DMX 500 (500 MHz) spectrometer, J values are given in Hz. Assignment of 1H NMR spectra could not be performed for the TTFs 6 and 15 due to isomeric mixtures. Mass spectra were recorded on a Finnigan SSQ 7000 or a Bruker Reflex III MALDI-MS instrument. The starting materials are commercially available except for 7h and 7i, which were obtained via bromination of the corresponding 1- and 2-naphthylacetic acids. Piperidinium tetrathiotungstate (14) was synthesized according to a literature procedure.20 Where stated, the silica gel was deactivated with a 2 M HCl solution with subsequent drying at rt before use. THF was distilled from sodium/benzophenone, all other solvents were either distilled or were HPLC grade.Acknowledgements
Financial support from the Swedish Research Council as well as the Foundation for Strategic Research is gratefully acknowledged. The assistance of Anna Herland and Andreas Woldegiorgis is also gratefully acknowledged.References
- A. M. Kini, B. D. Gates, M. A. Beno and J. M. Williams, J. Chem. Soc., Chem. Commun., 1989, 169 RSC.
- L. B. Coleman, M. J. Cohen, D. J. Sandman, F. G. Yamagishi, A. F. Garito and A. J. Heeger, Solid State Comm., 1973, 12, 1125 CrossRef CAS.
- P. Bernier, S. Lefrant and G. Bidan, Advances in Synthetic Metals; Twenty Years of Progress in the Science and Technology, Elsevier, Amsterdam, 1999 Search PubMed.
- (a) F. Garnier, G. Horowitz, X. Peng and D. Fichou, Adv. Mater., 1990, 2, 592 CrossRef CAS; (b) F. Garnier, R. Hajlaoui, A. Yassar and P. Srivastava, Science, 1994, 265, 1684; (c) T. Izumi, S. Kobashi, K. Takimiya, Y. Aso and T. Otsubo, J. Am. Chem. Soc., 2003, 125, 5286 CrossRef; (d) T. Otsubo, Y. Aso and K. Takimiya, J. Mater. Chem., 2002, 12, 2565 RSC.
- G. G. Abashev, E. V. Shklyaeva and V. S. Russkikh, Russ. J. Org. Chem., 1997, 33, 1652 CAS.
- (a) A. Ulman, An Introduction to Utrathin Films, Academic Press 1991 Search PubMed; (b) A. Ulman, J. Am. Chem. Soc., 1990, 112, 7083 CrossRef CAS; (c) A. Ulman, J. Am. Chem. Soc., 1991, 113, 4121 CrossRef; (d) A. Ulman, J. Am. Chem. Soc., 1991, 113, 5866 CrossRef; (e) A. Ulman, J. Am. Chem. Soc., 1991, 113, 6136; (f) A. Ulman, Adv. Mater., 1990, 2, 573 CAS; (g) G. Horowitz, F. Deloffre, B. Servet, S. Ries and P. Alnot, J. Am. Chem. Soc., 1993, 115, 8716 CrossRef CAS; (h) M. Reed and J. M. Tour, Sci. Am., 2000, 282(6), 86 Search PubMed.
- J. H. Schön, Adv. Mater., 2002, 14, 323 CrossRef CAS.
- C. M. Yip and M. D. Ward, Langmuir, 1994, 10, 549 CrossRef CAS.
- A. J. Moore, L. M. Goldenberg, M. R. Bryce, M. C. Petty, J. Moloney, J. A. K. Howard, M. J. Joyce and S. N. Port, J. Org. Chem., 2000, 65, 8269 CrossRef CAS.
- G. Trippé, M. Oçafrain, M. Besbes, V. Monroche, J. Lyskawa, F. Le Derf, M. Sallé, J. Becher, B. Colonna and L. Echegoyen, New J. Chem., 2002, 26, 1320 RSC.
- (a) S. Liu, H. Liu, K. Bandyopadhyay, Z. Gao and L. Echegoyen, J. Org. Chem., 2000, 65, 3292 CrossRef CAS; (b) M. Á. Herranz, B. Colonna and L. Echegoyen, PNAS, 2002, 99, 5040 Search PubMed.
- H. Fujihara, H. Nakai, M. Yoshihara and T. Maeshima, Chem. Commun., 1999, 733 RSC.
- M. Dorogi, J. Gomez, R. Osifchin, R. P. Andres and R. Reifenberger, Phys. Rev. B, 1995, 52, 9071 CrossRef CAS.
- A. Souizi, A. Robert, P. Batail and L. Ouahab, J. Org. Chem., 1987, 52, 1611 CrossRef.
- (a) A. Souizi and A. Robert, Synthesis, 1982, 1059 CrossRef CAS; (b) M. Jørgensen, K. Lerstrup and K. Bechgaard, J. Org. Chem., 1991, 56, 5684 CrossRef.
- E. Klingsberg, J. Am. Chem. Soc., 1964, 86, 5290 CrossRef CAS.
- J. Hu and M. A. Fox, J. Org. Chem., 1999, 64, 4959 CrossRef CAS.
- Only starting material could be recovered from the reaction of either 13f or 13g with hexamethyldisilathiane.
- P. Dhar, N. Chidambaram and S. Chandrasekaran, J. Org. Chem., 1992, 57, 1699 CrossRef CAS.
- G. Schukat, A. M. Richter and E. Fanghänel, Sulfur Reports, 1987, 7, 155 Search PubMed; G. Schukat and E. Fanghänel, Sulfur Rep., 1993, 14, 245 Search PubMed.
- C. D. Bain, E. B. Troughton, Y.-T. Tao, J. Evall, G. M. Whitesides and R. G. Nuzzo, J. Am. Chem. Soc., 1989, 111, 321 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data for the new compounds. See http://www.rsc.org/suppdata/jm/b3/b310260b/ |
| This journal is © The Royal Society of Chemistry 2004 |