Photoinduced electron transfer in silica-supported self-assembled thin films containing a 1,4,5,8-naphthalenetetracarboxylic diimide and cytochrome c
Ivana B.
Campos
,
Iseli L.
Nantes
,
Flavio A.
Rodrigues
and
Sergio
Brochsztain
*
Centro Interdisciplinar de Investigação Bioquímica, Universidade de Mogi das Cruzes, Av. Dr. Cândido Xavier de Almeida Souza, 200, Mogi Das Cruzes, SP 08780-911, Brazil. E-mail: brochsztain@umc.br; Fax: (55)
(11) 47987068; Tel: (55)
(11) 47987102
First published on 27th November 2003
Abstract
Self-assembled thin films containing an inner monolayer of N,N′-bis(2-phosphonoethyl)-1,4,5,8-naphthalenetetracarboxylic diimide (PNDI) and an outer monolayer of the cationic protein cytochrome c (cyt c) were prepared on the surface of non-porous silica gel particles. Silica particles modified with PNDI alone or with cyt c alone were also prepared for comparison. The materials were characterized by elemental and thermogravimetric analysis, as well as UV-visible absorption, fluorescence and infrared spectroscopies. Binding of PNDI to the silica resulted in a low density, sub-monolayer coverage, in contrast to cyt c adsorption which gave a rather compact protein layer. The loading of cyt c found in the new materials (ca. 12 µmol g−1) was higher than that found in the literature for porous silica gel. Irradiation of the materials at the imide absorption band resulted in the reduction of the cyt c heme iron, showing that the protein retained its activity upon binding to the modified silica gel surface. The proposed mechanism involves electron transfer from a photogenerated imide radical anion to the heme iron of the protein in the adjacent layer.
Introduction
1,4,5,8-Naphthalenetetracarboxylic diimides (NDI) are quite stable aromatic molecules that have attracted the attention of researchers from different areas in recent years. The interest in NDI arises mainly from their remarkable photophysical,1,2 photochemical3 and electrochemical4 properties. NDI are easily reduced, giving rise to stable imide radical anions,5 making them suitable building blocks for the design of new materials. Examples of NDI-containing materials include conducting polymers,5 field-effect transistors6 and model systems for photosynthesis.7 It is also worth mentioning the potential applications of NDI in bioorganic materials, since they have the ability to form intercalation complexes with DNA.8 Irradiation of these complexes leads to the photoinduced cleavage of DNA,9 placing NDI as strong candidates for the photodynamic therapy of cancer.In order to fully exploit the potential of NDI in materials chemistry, it is important to incorporate the molecules into organized media. In this regard, our group has been working on the construction of NDI-containing self-assembled thin films.10,11 For this purpose, we synthesized N,N′-bis(2-phosphonoethyl)-1,4,5,8-naphthalenetetracarboxylic diimide (PNDI), which is substituted with phosphonic acid groups, allowing the incorporation of the imide in self-assembled films using the well-known zirconium phosphonate (ZP) technique.12 Using this approach, we have been able to build films containing up to 20 layers of PNDI on flat surfaces, such as quartz, silicon and gold.10,11 Thin films built by the ZP technique are rather stable, which represents a great advantage over the NDI films grown by the Langmuir–Blodgett (LB) technique, as reported by other authors.13,14 Furthermore, the ZP approach allows the construction of self-assembled films on non-planar surfaces, such as silica gel particles,15 which is not possible with LB films.
We have recently reported the preparation and photophysical properties of PNDI self-assembled monolayers immobilized on the surface of zirconated silica gel.16 In that study, however, porous silica gel was employed. The present paper describes the construction of novel self-assembled materials, consisting of a monolayer of PNDI covered with a monolayer of the cationic protein cytochrome c (cyt c), supported on the surface of non-porous silica gel. Monolayers of cyt c adsorbed directly on the non-porous silica support, without the imide layer, were also prepared as reference materials.
Cyt c is a small redox protein that participates in the mitochondrial electron transfer (ET) chain. It contains a heme group whose central iron atom can be reversibly reduced from the Fe(III) to the Fe(II) state.17 The immobilization of cyt c on a variety of solid substrates,18,19 including mesoporous silica gel,20,21 has been the focus of literature reports, since immobilized redox proteins have important applications as biosensors.22 However, there has been no report, to our knowledge, on the immobilization of cyt c using non-porous silica gel.
The materials described here display very interesting photochemical properties. Irradiation of the films at the imide absorption band resulted in the conversion of cyt c to its reduced form, bringing evidence for the occurrence of interlayer ET from the imide layer to the protein layer. The cyt c remained indefinitely in the Fe(II) state after the irradiation, resulting in a very long-lived charge separated state, which shows that these materials are potentially useful as artificial photosynthetic devices.
Experimental
Materials
Silica gel Cab-O-Sil L-90 (supplier data: BET surface area 90 m2 g−1, average particle diameter 20 nm) was a gift fom Cabot Corporation, and was used as received. Zirconyl chloride octahydrate and collidine were purchased from Aldrich and used as received. Phosphoryl chloride (Aldrich) was distilled prior to use. PNDI was synthesized by a method previously reported by our group.2 Cytochrome c (from horse heart) was purchased from Sigma. Acetonitrile (Mallinckrodt) was dried by distillation under P2O5. All aqueous solutions were prepared with deionized water (Barnstead easypure RF system).Instruments
Samples were stirred in a model C24 Incubation Shaker (New Brunswick Scientific). Centrifugation was performed either in a Himac CR 21E centrifuge or in a Himac CF 15R microcentrifuge (Hitachi). Elemental analysis (C,H,N) of the samples was done by the Chemical Analysis Laboratory, at the Chemistry Institute, Universidade de São Paulo. Thermogravimetric analyses (TGA) were performed using a model TGA 7 Thermogravimetric Analyzer (Perkin Elmer). Typically, samples were held at 120 °C for 1 min to remove water, then heated up to 900 °C at a rate of 20 °C min−1, under nitrogen atmosphere. UV-visible absorption spectra were recorded in a Shimadzu Multi-Spec 1501 spectrophotometer. Emission spectra were taken with a Hitachi F 2500 Fluorescence Spectrophotometer. Infrared spectra were recorded with the samples in KBr pellets, using a FTIR Perkin-Elmer Spectrum One apparatus. Irradiation of the samples was performed at 365 nm, with a 4 W compact lamp, model UVGL-25 (UVP).Methods
Results
Preparation of the materials
| Material | % C | % H | % N |
|---|---|---|---|
| a Commercial Cab-O-Sil, as received from the supplier. b After exhaustive washing with water. | |||
| (a) untreated silicaa | 0.23 | 0.16 | 0.06 |
| (b) zirconated silica + PNDI (Sil/PNDI)b | 2.01 | 0.31 | 0.25 |
| (c) untreated silica + PNDIb | 0.33 | 0.10 | 0.05 |
| (d) zirconated silica + PNDI + cyt c (Sil/PNDI/Cyt)b | 8.28 | 1.22 | 1.93 |
| (e) untreated silica + cyt c (Sil/Cyt)b | 7.22 | 1.06 | 2.02 |
Characterization of the materials
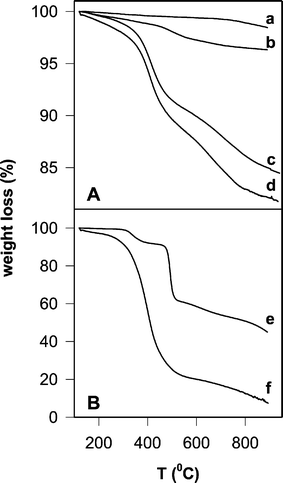 | ||
| Fig. 1 (A) TGA analysis of silica gel samples: (a) commercial silica, (b) Sil/PNDI, (c) Sil/Cyt, (d) Sil/PNDI/Cyt. (B) TGA analyses of the coating molecules in the crystalline state: (e) PNDI, (f) cyt c. | ||
The observed thermal process for pure PNDI (Fig. 1B) can be attributed to the combustion of the organic portion of the molecule, leaving behind a residual amount of inorganic phosphorus-containing degradation products (45% in weight in the case of pure PNDI), since other NDI without phosphonate substituents (e.g.: N,N′-bisphenyl-1,4,5,8-naphthalenetetracarboxylic diimide) showed a weight loss of more than 90% in TGA analysis, according to literature results.24
TGA analysis of Sil/PNDI/Cyt (Fig. 1A) showed a weight loss of 18.3%, which represents a further 14.3% loss relative to the precursor material Sil/PNDI, clearly demonstrating the binding of the protein onto the imide layer. TGA analysis of Sil/Cyt is also shown (15.5% weight loss), confirming that cyt c was also adsorbed on the non-modified silica, in agreement with the elemental analysis data. Most of the weight loss in Sil/Cyt and Sil/PNDI/Cyt occurred within the temperature range 330–490 °C, which was approximately the range observed in the TGA of the crystalline protein (Fig. 1B). The thermal decomposition of the imide in Sil/PNDI/Cyt was not clearly seen because the corresponding weight loss was small compared to the (superimposed) weight loss due to protein decomposition.
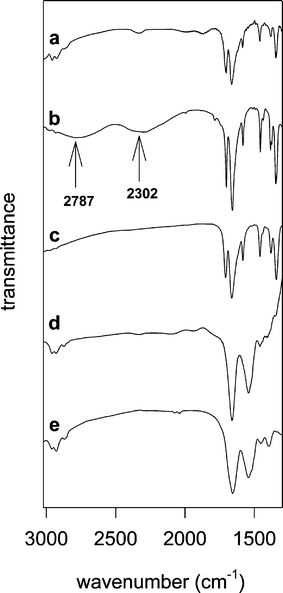 | ||
| Fig. 2 FTIR spectra in KBr pellets: (a) Sil/PNDI, (b) crystalline PNDI, (c) bulk PNDI/Zr material (precipitated from solution), (d) Sil/PNDI/Cyt, (e) crystalline cyt c. Contributions from the silica to the spectra were subtracted in (a) and (d). | ||
The main difference between the spectra of free and silica-supported PNDI is the absence of the broad bands at 2787 and 2302 cm−1 in the spectrum of the bound imide. These bands are attributable to the O–H stretch mode of the phosphonic acid groups.25 As a comparison, the spectrum of a bulk zirconium phosphonate solid, obtained by precipitation of PNDI from solution with a Zr salt, is shown in Fig. 2 (trace c). The absence of the hydroxyl bands was also observed in this case, and can be attributed to the exchange of the hydrogen atoms by zirconium cations in solid zirconium phosphonates.26 These results suggest that PNDI was bound to the zirconium layer in Sil/PNDI through specific interactions with the phosphonate groups.
Fig. 2 also shows the infrared spectrum of Sil/PNDI/Cyt (trace d). The spectrum is very similar to that of crystalline cyt c (trace e), showing clearly the amide I and amide II bands characteristic of proteins (1657 and 1543 cm−1, respectively). The bands of PNDI are not visible in the spectrum of Sil/PNDI/Cyt because the carbonyl groups from the protein are by far more abundant than imide carbonyls in this material (see discussion), and therefore the protein bands predominate.
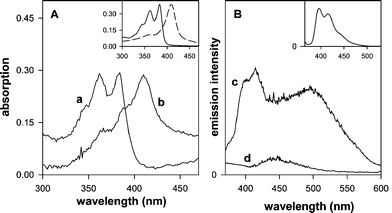 | ||
| Fig. 3 (A) Absorption spectra of modified silica gel samples suspended in water: (a) Sil/PNDI, (b) Sil/PNDI/Cyt. The spectra were baseline corrected to compensate for the scattering due to the silica. Inset: absorption spectra of PNDI (1.8 × 10−5 M) (—) and of cyt c (3.9 × 10−6 M) (- - -) in aqueous solution. (B) Emission spectra of the modified silica gel suspended in water: (c) Sil/PNDI, (d) Sil/PNDI/Cyt. Excitation wavelength = 330 nm. Inset: emission spectrum of PNDI (1.8 × 10−5 M) in aqueous solution. | ||
It should be noticed that the presence of PNDI in the Sil/PNDI/Cyt material could not be detected clearly by any of the methods described above, due to the presence of the cyt c layer. This could indicate that the imide was either removed during the washing steps in the preparation of Sil/PNDI/Cyt or decomposed by the presence of the protein. In order to eliminate this possibility, digestion of the material with hydrofluoric acid was carried out, resulting in a solution containing both PNDI and cyt c (according to UV-Vis analysis) in the expected ratio, as determined from the elemental analysis and TGA data. This result is very important, confirming that the imide layer was still present in Sil/PNDI/Cyt after the deposition of the protein layer.
Irradiation of the modified silica
When aqueous suspensions of Sil/PNDI/Cyt were irradiated at 365 nm, pronounced changes occurred in the absorption spectra of the samples (Fig. 4). A red shift in the Soret band and the appearance of new absorption bands at 530 and 549 nm were observed. These spectral changes are characteristic of the reduction of cyt c from the Fe(III) to the Fe(II) state.17 No changes were observed when cyt c alone, either in solution or adsorbed on silica (Sil/Cyt), was irradiated under the same conditions without the presence of PNDI. Irradiation of the silica coated with PNDI alone did not cause any observable change in the spectra as well. The observed phenomenon can therefore be attributed to a photoinduced electron transfer from the imide to the protein heme group (see discussion). After the irradiation, the protein remained indefinitely in the reduced form, showing that back electron transfer did not occur in the films at an appreciable rate. Remarkably, the photochemical reaction occurred even in the presence of atmospheric oxygen.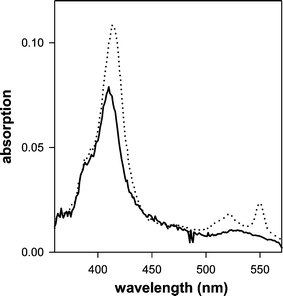 | ||
| Fig. 4 Absorption spectra of an aqueous suspension of Sil/PNDI/Cyt, before irradiation (—) and after 15 min of irradiation at 365 nm (· · ·). The spectra were baseline corrected to compensate for the scattering due to the silica. | ||
Discussion
Structure of the new materials
The elemental analysis and TGA data of the modified silica materials allow the calculation of surface coverages, as shown in Table 2 (the results are presented in both weight % and µmol g−1 units). A satisfactory agreement was found between the surface coverages obtained by the two different methods, especially in the case of Sil/PNDI. Taking into account the given specific surface area for the silica gel employed in this study (90 m2 g−1, according to the manufacturer), the footprints of the molecules on the surface could be estimated, and are also displayed in Table 2.| Sample | Amount of adsorbed PNDI or cyt c | Area per molecule/Å2 | |
|---|---|---|---|
| weight % | µmol g−1 | ||
| a The average of the values obtained using the C, H and N contents (or C and H only in the case of Sil/PNDI/Cyt) was used for calculation. b The weight loss obtained for the blank (blank = untreated silica for Sil/PNDI and Sil/Cyt; Sil/PNDI for Sil/PNDI/Cyt) was discounted from the total weight loss before calculation. c Data relative to modification with PNDI. d Data relative to modification with cyt c. | |||
| Sil/PNDIc | 3.9 (4.2) | 85.0 (90.0) | 167 (158) |
| Sil/Cytd | 13.3 (15.1) | 12.4 (14.4) | 1214 (1040) |
| Sil/PNDI/Cytd | 12.7 (15.5) | 11.8 (14.9) | 1276 (1010) |
Let us next consider the arrangement of the PNDI molecules at the interface of Sil/PNDI. The excimer-like band observed in the emission spectrum of this material (Fig. 3B) is similar to that observed with NDI ground state aggregates,1 suggesting the presence of surface aggregates of π-stacked imide molecules, as shown in Fig. 5A. The formation of surface islands has already been observed in the case of PNDI thin films on flat surfaces.10 The observation of monomer emission in the spectrum of Sil/PNDI indicates, on the other hand, that isolated PNDI molecules are also present on the surface. In this case, two different binding modes are possible, namely PNDI molecules attached to the surface by only one phosphonate group, as in Fig. 5B, or imide molecules anchored with both phosphonate groups, as in Fig. 5C. The disappearance of the vibrational modes associated with the O–H stretch in the FTIR spectrum of Sil/PNDI (Fig. 2) might be an indication that most isolated molecules are bound with both sides, although a conclusive assignment is not possible based only on the present results.
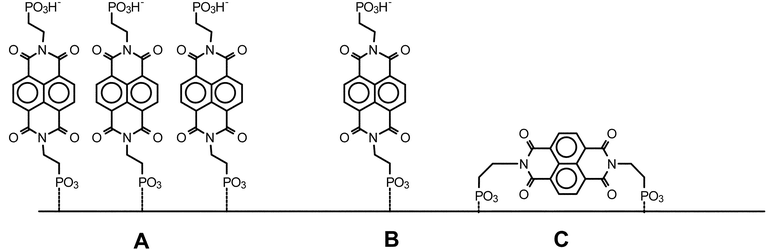 | ||
| Fig. 5 Different arrangements possible for the binding of PNDI to the zirconated silica gel surface. (A) Islands of π-stacked molecules. (B) Isolated molecules attached with one phosphonate group. (C) Isolated molecules attached with both phosphonate groups. | ||
Attempts to grow a second layer of PNDI on the first imide layer, by exposing Sil/PNDI to a Zr solution followed by a PNDI solution, resulted in the incorporation of a much smaller amount than that observed in the deposition of the first layer. This observation is consistent with a significant amount of PNDI molecules being bound with both ends (Fig. 5C), as mentioned above, since exposed phosphonate groups are needed for the growth of a second layer.
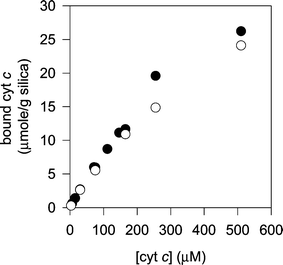 | ||
| Fig. 6 Binding isotherms for the adsorption of cyt c to non-modified silica gel (filled circles) and to PNDI-modified silica gel (open circles). | ||
This lack of selectivity can be explained by the low density of imide molecules on the PNDI layer, leaving exposed large areas of the bare silica gel. In this regard, it is most likely that these exposed areas were not modified during the priming treatment (i.e., phosphorylation and zirconation), which also explains why the PNDI molecules did not bind to these regions. Thus, the binding of cyt c should be dominated by interactions with the silica surface, rather than interactions with the phosphonates or other functional groups of PNDI (Fig. 7). Electrostatic attraction between the positively charged cyt c and the negative charges of the deprotonated silanols on the silica surface probably plays an important role in the binding, as reported by other authors in the case of mesoporous silica.27 It is interesting to compare the present results with literature reports showing a rather low affinity for the adsorption of bovine serum albumin, a negatively charged protein, to silica gel surfaces.28
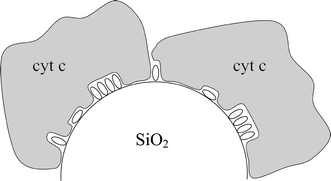 | ||
| Fig. 7 Schematic structure of Sil/PNDI/Cyt, showing the intermingling of the internal PNDI layer (small ellipses) and the external cyt c layer. | ||
Regarding the density of cyt c in the protein layer, a molecular area of about 11 nm2 per molecule of protein was estimated for both Sil/PNDI/Cyt and Sil/Cyt materials (Table 2). This value is close to the cross sectional area of 9.3 nm2 estimated for cyt c,29 indicating that a compact monolayer of cyt c was formed, regardless of the presence or not of the imide layer. It is worth mentioning that the loading of cyt c in these materials was superior to the values achieved by other authors. Deere et al., for example, obtained a maximum loading of about 8 µmol g−1 for cyt c adsorbed on mesoporous silica,21 which is lower than the values reported here (11–14 µmol g−1) with the non-porous silica. These differences can be explained by the low accessibility of the protein to the internal surface of the pores, a problem not encountered in the case of the non-porous silica, where all the surface is external.
It can be noticed in Fig. 6 that the binding isotherms tend to reach a plateau at a surface coverage of about 25 µmolg−1, which is nearly double the coverage estimated by elemental analysis and TGA in Table 2. This difference probably arises from the different experimental conditions employed, since the samples Sil/PNDI/Cyt and Sil/Cyt were exhaustively washed with water during the preparation, in order to remove weakly adsorbed protein. The isotherms in Fig. 6 were produced without washing the silica, which could leave a second layer of weakly adsorbed cyt c deposited on the first protein layer. If the weakly adsorbed protein is to be taken into account, the loading of cyt c obtained in this work is up to four times that reported with mesoporous silica.21
Photochemical behaviour of the new materials
Based on previous knowledge on the photochemical behaviour of NDI derivatives,2,3,30,31 a plausible mechanism for the observed photoreduction of cyt c in Sil/PNDI/Cyt can be proposed, as shown in Scheme 1. Excitation of the imide at 365 nm leads to the lowest singlet state (step 1), followed by intersystem crossing to the triplet state (step 2). Efficient intersystem crossing has been observed for other NDI derivatives, with near unity quantum yield.3,30 Therefore, the formation of 3(PNDI)* in the films is quite expected. The triplet state of PNDI can accept an electron from any donor present in the surroundings,3,30 generating an imide radical anion (step 3). Radical anions of NDI have been well characterized, and are known to be relatively stable species.5,7 The radical anion can then transfer an electron to cyt c heme iron in the adjacent layer (step 4), leading to the reduction of the protein, thus explaining the results shown in Fig. 4. The donor in step 3 is most likely a PNDI molecule in the ground state (self-quenching ET), as proposed by other authors.3,31 The fluorescence results (Fig. 3) show that some of the imide molecules are stacked on the surface (see Fig. 5), which would favour the intralayer ET from a ground state PNDI to a 3(PNDI)*. Another possibility is that an amino group from the protein acts as the electron donor in Scheme 1. We are presently investigating the photochemical mechanism in more detail, including studies in homogeneous solutions.32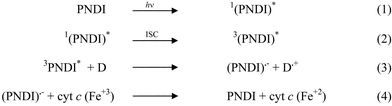 | ||
| Scheme 1 Proposed mechanism for the photochemical reaction between PNDI and cyt c in Sil/PNDI/Cyt. | ||
Conclusions
We have shown that rather stable monolayers of NDI can be grown on silica surfaces using zirconium phosphonate chemistry. The films are quite insoluble, and do not desorb from the surface even in the presence of polar solvents like water. In addition, we have shown that a layer of the protein cyt c can be readily adsorbed onto the imide layer, giving rise to photoactive materials. These materials undergo permanent photoinduced electron transfer, generating cyt c Fe(II). The observed photochemical behaviour of Sil/PNDI/Cyt shows that cyt c retains its redox activity when adsorbed to the silica surface, which is a highly desirable property for any possible application of this system. The system studied here has potential applications for solar energy storage devices, as well as in the field of biosensors and in the sensitization of photodynamic therapy for cancer.Acknowledgements
The authors are grateful to the Brazilian agency FAPESP for financial support. Grant numbers: IC 01/12033-3 (I.B.C.), JP 99/07114-2 (S.B.), JP 98/13659-9 (I.L.N.) and JP 98/09644-6 (F.A.R.). Cabot Corporation is greatly acknowledged for the kind gift of a sample of Cab-O-Sil L-90. S.B. and F.A.R. wish to thank Fundação de Amparo ao Ensino e Pesquisa (FAEP) for a research fellowship.References
- T. C. Barros, S. Brochsztain, V. G. Toscano, P. Berci-Filho and M. J. Politi, J. Photochem. Photobiol. A:Chem., 1997, 111, 97–104 CrossRef CAS.
- M. A. Rodrigues, S. Brochsztain, T. C. Barros, M. S. Baptista and M. J. Politi, Photochem. Photobiol., 1999, 70, 35–39 CAS.
- B. M. Aveline, S. Matsugo and R. W. Redmond, J. Am. Chem. Soc., 1997, 119, 11785–11795 CrossRef CAS.
- A. Viehbeck, M. J. Goldberg and C. A. Kovac, J. Electrochem. Soc., 1990, 137, 1460–1466 CAS.
- L. L. Miller and K. R. Mann, Acc. Chem. Res., 1996, 29, 417–423 CrossRef CAS.
- H. E. Katz, A. J. Lovinger, J. Johnson, C. Kloc, T. Siegrist, W. Li, Y. Y. Lin and A. Dobabalapur, Nature, 2000, 404, 478–481 CrossRef CAS.
- S. R. Greenfield, W. A. Svec, D. Gosztola and M. R. Wasielewski, J. Am. Chem. Soc., 1996, 118, 6767–6777 CrossRef CAS.
- R. S. Lokey, Y. Kwok, V. Guelev, C. J. Pursell, L. H. Hurley and B. L. Iverson, J. Am. Chem. Soc., 1997, 119, 7202–7210 CrossRef CAS.
- M. E. Nuñez, K. T. Noyes, D. A. Gianolio, L. W. McLaughlin and J. K. Barton, Biochemistry, 2000, 39, 6190–6199 CrossRef CAS.
- M. A. Rodrigues, D. F. S. Petri, M. J. Politi and S. Brochsztain, Thin Solid Films, 2000, 371, 109–113 CrossRef CAS.
- S. Brochsztain, M. A. Rodrigues, G. J. F. Demets and M. J. Politi, J. Mater. Chem., 2002, 12, 1250–1255 RSC.
- G. Cao, H. G. Hong and T. E. Mallouk, Acc. Chem. Res., 1992, 25, 420–427 CrossRef CAS.
- V. Cammarata, L. Atanasoska, L. L. Miller, C. J. Kolaskie and B. J. Stallman, Langmuir, 1992, 8, 876–886 CrossRef CAS.
- D. E. Lynch, D. G. Hamilton, N. J. Calos, B. Wood and J. K. M. Sanders, Langmuir, 1999, 15, 5600–5605 CrossRef CAS.
- H. G. Hong, D. D. Sackett and T. E. Mallouk, Chem. Mater., 1991, 3, 521–527 CrossRef CAS.
- M. A. Rodrigues, M. P. Bemquerer, M. J. Politi, S. Brochsztain, M. T. M. Miranda and M. S. Baptista, Chem. Lett., 2002, 604–605 CrossRef CAS.
- I. L. Nantes, M. R. Zucchi, O. R. Nascimento and A. Faljoni-Alario, J. Biol. Chem., 2001, 276, 153–158 CrossRef CAS.
- Y. Lvov, K. Ariga, I. Ichinose and T. Kunitake, J. Am. Chem. Soc., 1995, 117, 6117–6123 CrossRef CAS.
- P. L. Edmiston, J. E. Lee, S. S. Cheng and S. S. Saavedra, J. Am. Chem. Soc., 1997, 119, 560–570 CrossRef CAS.
- L. Washmon-Kriel, V. L. Jimenez and K. J. Balkus, J. Mol. Catal. B: Enzym.., 2000, 10, 453–469 CrossRef CAS.
- J. Deere, E. Magner, J. G. Wall and B. K. Hodnett, Chem. Commun., 2001, 465–465 RSC.
- I. Willner and E. Katz, Angew. Chem., Int. Ed., 2000, 39, 1180–1218 CrossRef.
- P. Kohli and G. J. Blanchard, Langmuir, 2000, 16, 695–701 CrossRef CAS.
- D. Uzun, M. E. Ozser, K. Yuney, H. Icil and M. Demuth, J. Photochem. Photobiol. A: Chem., 2003, 156, 45–54 CrossRef CAS.
- L. C. Thomas, Interpretation of the Infrared Spectra of Organophosphorus Compounds, Heyden, London, 1974, ch. 3 Search PubMed.
- H. C. Yang, K. Aoki, H. G. Hong, D. D. Sackett, M. F. Arendt, S. L. Yau, C. M. Bell and T. E. Mallouk, J. Am. Chem. Soc., 1993, 115, 11855–11862 CrossRef CAS.
- J. Deere, E. Magner, J. G. Wall and B. K. Hodnett, J. Phys. Chem. B, 2002, 106, 7340–7347 CrossRef CAS.
- S. Robinson and P. A. Williams, Langmuir, 2002, 18, 8743–8748 CrossRef CAS.
- T. Takano, O. B. Kallai, R. Swanson and R. E. Dickerson, J. Biol. Chem., 1973, 248, 5234–5255 CAS.
- S. Green and M. A. Fox, J. Phys. Chem., 1995, 99, 14752–14757 CrossRef CAS.
- J. E. Rogers, S. J. Weiss and L. A. Kelly, J. Am. Chem. Soc., 2000, 122, 427–436 CrossRef CAS.
- I. B. Campos, I. L. Nantes, M. J. Politi and S. Brochsztain, Photochem. Photobiol. Search PubMed , submitted.
| This journal is © The Royal Society of Chemistry 2004 |