Synthesis of conjugated oligomers and polymers: the organometallic way
Francesco Babudriab, Gianluca M. Farinolaa and Francesco Naso*ab
aDipartimento di Chimica, Università degli Studi di Bari, via Orabona, 4, Bari, I-70126, Italy
bCNR ICCOM, Dipartimento di Chimica, Università degli Studi di Bari, via Orabona, 4, I-70126, Italy. E-mail: naso@area.ba.cnr.it; Fax: 0039 080 5442924; Tel: 0039 080 5442073
First published on 2nd December 2003
Abstract
Results of special interest in the field of organometallic chemistry directed towards the synthesis of conjugated oligomers and polymers are reviewed. Methodologies widely employed in the synthesis of well-defined molecules and based on the transition metal catalyzed coupling reactions of various organometallic (B, Sn, Si, Mg and Zn) reagents have been more recently extended to build up poly- or oligo-meric conjugated molecular architectures. Although the focus of the present work is on transition metal cross-coupling or homo-coupling processes, other useful organometallic routes, such as metathesis reactions and acetylene polymerization, have also been dealt with. Owing to their higher versatility with respect to the non-organometallic processes commonly used for the synthesis of these materials, the approaches presented allow the realization of more complex molecular structures, which are nowadays required in many electronic and optoelectronic applications. The use of organometallic strategies has been discussed from a synthetic organic chemist's point of view, and advantages and limitations of the different methodologies reviewed have been highlighted. Some attention has also been paid to the properties of the resulting materials and to their dependence on the procedure followed and the framework obtained.
 Francesco Babudri | Francesco Babudri was born in Bari (Italy) in 1953 and received his “Laurea” in Chemistry from the University of Bari in 1976. He started his career as a Researcher in 1978 at the Department of Chemistry of the same University, working with Professor S. Florio in the field of organometallic chemistry. After a stay at the “Laboratoire de Chimie” of the “Ecole Normale Superieure” in Paris (1988) with Professor P. Sinaÿ, he joined the research group of Professor F. Naso at the University of Bari, working in the field of organometallic chemistry applied to the synthesis of polyunsaturated compounds. Currently, he is Professor of Organic Chemistry at the same University. His main research interests focus on polymer chemistry, materials, and synthetic strategies. |
 Gianluca Farinola | Gianluca M. Farinola was born in Bari (Italy) in 1968. He received his “Laurea” in Chemistry from the University of Bari in 1992, and his Ph.D. in Chemical Sciences in 1996, working with Professor F. Naso. In 1996 he started his career as a Researcher and, from 2002, he has been Associate Professor of Organic Chemistry at the University of Bari. His research interests deal with the synthesis and properties of polyconjugated polymeric and molecular materials for photonics and electronics. In 2003 he was awarded the Ciamician Medal of the Italian Chemical Society for young organic chemists. |
 Francesco Naso | Francesco Naso was born in Rosarno (Italy) in 1937 and received his “Laurea” in Industrial Chemistry from the University of Bologna in 1961, working with Professor G. Modena. After a stay (1962–63) at the University of Washington, Seattle, with Professor Y. Pocker, he moved to the University of Bari, where in 1975, after holding several intermediate positions, he became Professor of Organic Chemistry. From 1984 to 2000, he was Director of the C.N.R. Centre on “Metodologie Innovative di Sintesi Organiche”, a centre which, recently, has merged into the “Istituto di Chimica dei Composti Organometallici”. He is currently heading up the Bari section of this C.N.R. Institute. The scientific contributions made early in his career dealt with mechanistic and stereochemical features of fundamental organic reactions (e.g. nucleophilic vinylic substitutions and olefin or acetylene forming eliminations). His current research interests include organometallic chemistry, enantioselective processes, and synthesis of polyconjugated materials with electro-optical properties. |
1. Introduction
Organic polymers have been widely employed in electronic device fabrication mainly for their insulating properties. Starting from 1977, the role of some organic polymers and oligomers1 in the electronic industry has been continuously reconsidered in a new light as potential substitutes of conventional inorganic conductors and semiconductors in a great variety of applications.2 Indeed, the pioneering work of Shirakawa, MacDiarmid, Heeger and coworkers allowed thin films of polyacetylene to be obtained,3 a new organic polymer discovered by Natta in 1958,4 which exhibited a relatively high electrical conductivity by “doping” with bromine or iodine.5 This fundamental discovery opened the way to the development of a wide range of organic polyconjugated materials, with different structural frameworks based upon aromatic, heteroaromatic, vinylic or acetylenic π-systems (Fig. 1).Among the great variety of applications developed during the last two decades, PPV, PPE and PPP polymers have been employed in the fabrication of electroluminescent devices,6 plastic lasers,7 and photovoltaic cells.8 Furthermore, optical non-linearities of conjugated polymers offer potential applications in electro-optical and all-optical devices, such as digital optical switches, light modulators, logic gates, and high-density optical data storage devices.9 New molecular architectures are continuously designed with the aim of improving the performance of electrical and electrooptical devices based upon organic materials and, nowadays, the development of synthetic processes for realizing them represents a challenge for the organic chemist interested in this field. Each class of conjugated polymers has its own methodology of synthesis, characterized by variable flexibility in modifying the molecular structure of the material and compatibility with various functional groups. PPVs are generally prepared by the elimination reaction of non-conjugated precursor polymers (the Wessling route,10 or the Gilch route,11Scheme 1) which is amenable (particularly the Gilch process) to scale up to kilogram production.12
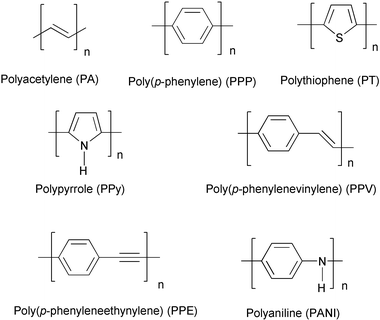 | ||
| Fig. 1 Representative structures of some classes of conjugated polymers. | ||
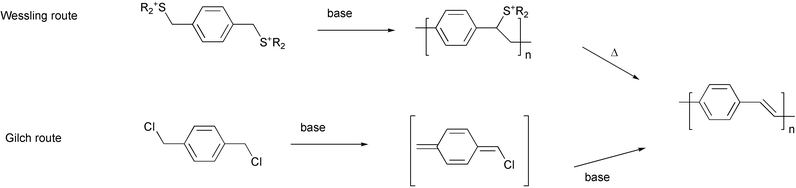 | ||
| Scheme 1 | ||
However, the conjugated system of these materials may present structural defects such as carbonyl groups or head-to-head coupling in the case of Wessling route, or the so-called tolane-bisbenzyl defects in the Gilch route (Fig. 2).
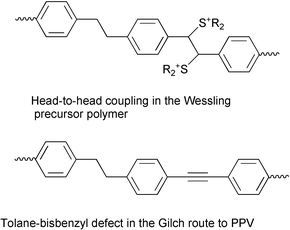 | ||
| Fig. 2 Possible structural defects in the precursor polymer routes. | ||
PPVs with large proportions of cis double bonds were also prepared by means of Wittig and related condensation reactions.13 A subsequent cis–trans isomerization step is required in order to obtain stereodefined polymers with an all-trans configuration of the vinylene units.13a
PPP polymers were prepared by the Kovacic's route14 (Scheme 2), but the reaction conditions are rather harsh for many functional groups.
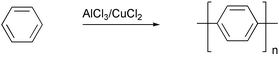 | ||
| Scheme 2 | ||
Oxidative polymerization with ferric chloride15 or electrochemical polymerization16 are useful synthetic methodologies to PTs, but they do not allow to obtain regioregular polymers from 3-monosubstituted thiophenes. PPy17 and PANI18 polymers are also prepared by oxidative methods.
Organometallic methodologies, widely exploited in the synthesis of well defined molecules, soon appeared very attractive for the preparation of polydisperse conjugated materials, especially when a high regio- and stereo-selectivity is required in building the polymeric backbone. Owing to the huge number of papers published in the last decade and dealing with the synthesis of conjugated polymers and oligomers by means of the most used organometallic approaches to unsaturated systems, an exhaustive review on this topic is beyond the scope of this article. Therefore, only an overview of the most recent and, in our opinion, most relevant results is presented below.
2. Arylation of alkenes (the Heck reaction)
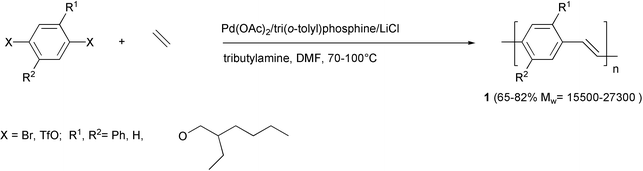
The synthesis of polymers with aromatic-vinylic structure can be performed by means of the palladium-catalysed coupling reaction of aryl halides with alkenes.19 This methodology, using monomers containing pre-formed carbon–carbon double bonds, avoids the presence of structural defects, which may occur in the alternative precursor polymer approach reported in the Introduction. A great variety of arylene-vinylene structures and the tunability of their electrooptical properties via altering the nature of the substituents on the conjugated system are allowed since the mild reaction conditions are compatible with many functionalities. Moreover, starting from suitable vinylic and aromatic monomers, copolymers with defined alternating structures can be obtained, whereas the precursor polymer route would permit only the preparation of random materials.The first PPV polymers 1 prepared by the Heck reaction are presented in Scheme 3.20 IR absorption in the region 962–970 cm−1 confirmed the trans configuration of the double bonds. However, significant amounts of 1,1-vinylidene moieties were found in the polymer backbone, which may affect the absorption, photoluminescence and electroluminescence spectra.
 | ||
| Scheme 3 | ||
The regular PPV copolymer 4, reported by Yu and coworkers,21 was obtained by a simple reaction of 1,4-divinyl benzene 2 with several dialkoxydiiodo benzenes 3 in DMF, in the presence of a tertiary amine, a triarylphosphine and a catalytic amount (2 mol%) of palladium acetate (Scheme 4). 1H and 13C NMR and FTIR spectral evidence suggested that the major configuration of the vinylene units is the trans form. Also in this case the coupling of the aryl halide with the α-vinyl position appeared to occur as a side reaction, and this was evidenced by the weak 1H NMR signals of the end β-protons.
 | ||
| Scheme 4 | ||
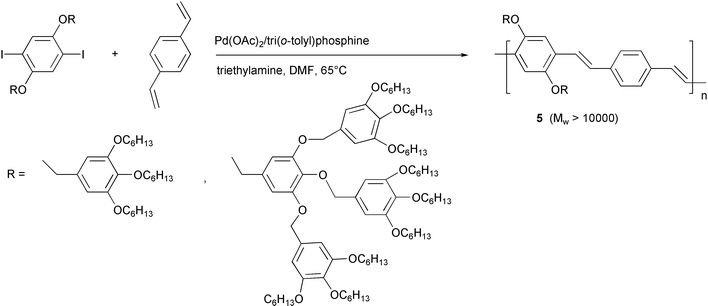
However, these drawbacks do not reduce the important role of the methodology in the synthesis of arylene-vinylene polymers. More recently, complex PPV structures 5 in which the simple linear alkyl or alkoxy chains are replaced by hyperbranched side groups (Scheme 5) have been prepared using the standard Heck reaction conditions.22 The reduction of solid state interchain interactions in this PPV derivative, induced by the presence of the bulky substituents, improves the electro- and photo-luminescence quantum efficiencies by reducing the aggregation quenching.
 | ||
| Scheme 5 | ||
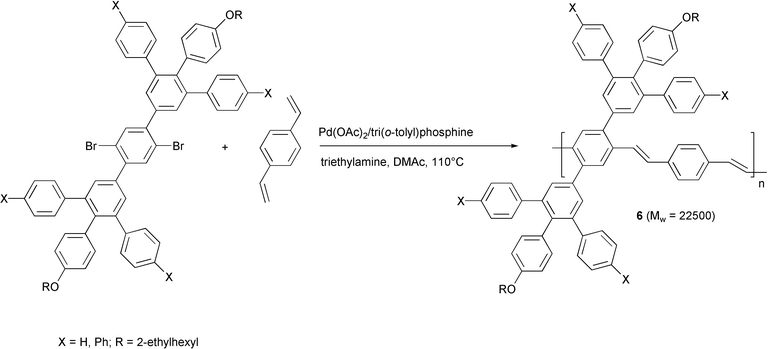
Similar results have been obtained in highly phenylated PPVs 6, prepared using similar experimental conditions (Scheme 6).23
 | ||
| Scheme 6 | ||
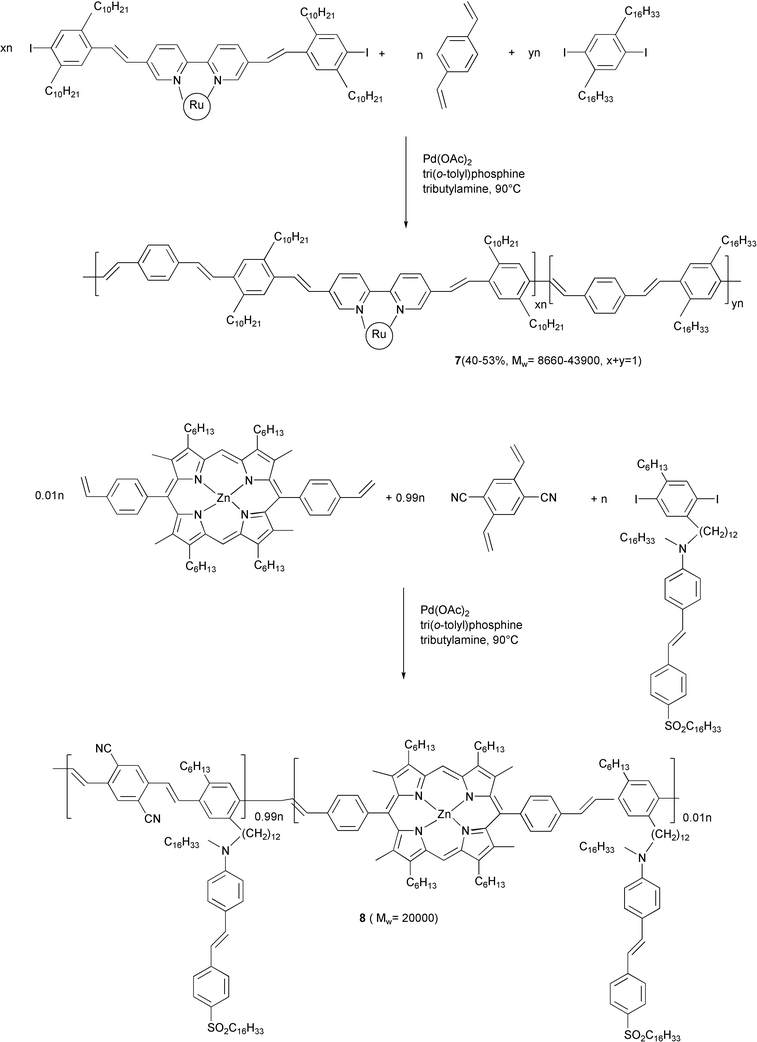
Polymers characterized by photoconductive24 (compound 7, Scheme 7) or photorefractive properties (compound 8)25 and containing transition metal complexes were also prepared by using this coupling reaction.
 | ||
| Scheme 7 | ||
PPV polymers represent the most promising organic semiconductors in the fabrication of electroluminescent devices (LEDs), and a careful balance of the rate of injection of positive and negative charge carriers, i.e. holes and electrons respectively, is a fundamental prerequisite for high quantum electroluminescence efficiencies, which markedly affect the performances of the devices. The introduction of oxadiazole units in the main chain of PPV polymers provided high electron affinity materials, with an improved electron injection which may be conveniently exploited in optimising the performance of light emitting devices. Two examples of this new kind of conjugated materials are reported in Scheme 8. The presence of meta linkages26 (9) or of silicon atoms27 (10) reduces the effective π-conjugation length in the main chain. As a consequence, a shift of the photoluminescence wavelength emission in the blue region occurs, and these materials appear very promising organic semiconductors for the fabrication of blue-emitting LEDs.
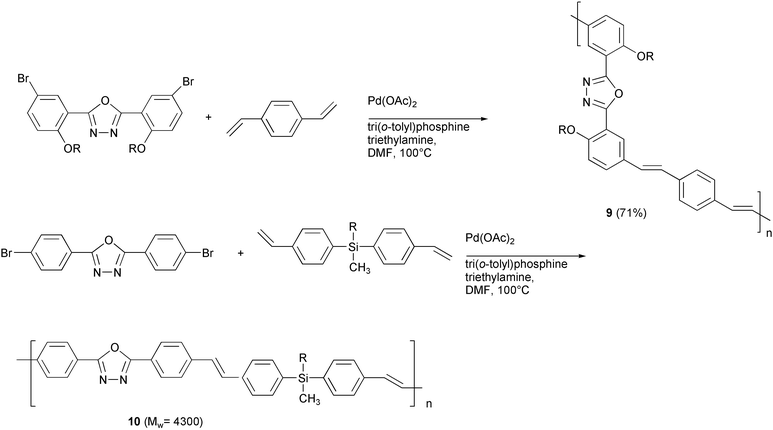 | ||
| Scheme 8 | ||
2.1 Non-conventional Heck arylation
Arenediazonium tetrafluoroborates have been employed as an alternative to aryl halides in a non-conventional arylation reaction of olefins. The greater reactivity of arenediazonium salts with respect to aryl bromides and iodides allows mild reaction conditions (room temperature to 80 °C) and shorter reaction times by using phosphine-free palladium catalyst, without the intervention of a base.28 In particular, aryl trimethylsilyl alkenes react with diazonium salts in a Heck type aryldesilylation process, as in the case of unsaturated silanes 1129 or 1330 (Scheme 9). It is worth noting that when the α-olefin 13 was used, the stilbene derivative 12 was obtained regio- and stereo-selectively.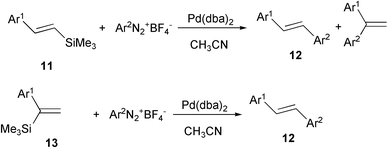 | ||
| Scheme 9 | ||
Pd-Catalyzed arylation and aryldesilylation processes occurred in the reaction of triethoxyvinylsilane 15 with arenebisdiazonium tetrafluoroborates 14 derived from benzidines to give low polydispersity poly(4,4′-biphenylenevinylene)s 16, using Pd(OAc)2 as the catalyst (Scheme 10).31
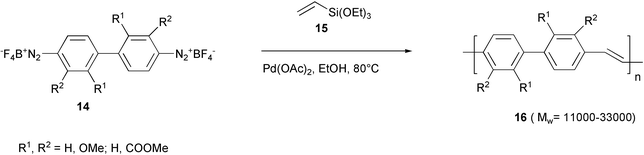 | ||
| Scheme 10 | ||
A double aryldesilylation strategy was applied to the synthesis of various new conjugated materials with interesting structural features, by reacting the α,α′-bis(trimethylsilyl) derivative 17 with various arenebisdiazonium tetrafluoroborates 18 (Scheme 11).32 The polymers 19b–d are characterized by a regular alternation of aromatic systems bearing electron-donating (octyloxy) and electron-withdrawing (carbonyl or sulfonyl) groups. These groups originate a strong electron polarisation in the conjugated system and, in turn, this may lead to enhanced non-linear optical properties. The reduced conjugation length of the π-system, due to the presence of methylene groups in the main chain of polymer 19e may have a special interest in the field of blue-emitting OLEDs, owing to the expected substantial shift of photoluminescence and electroluminescence towards shorter wavelengths.
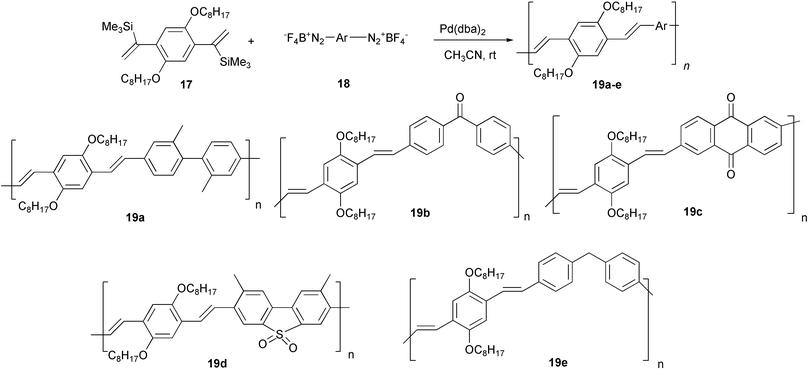 | ||
| Scheme 11 | ||
Conjugated monodisperse oligomers play a key role in the study of the properties of organic semiconductors, since a detailed knowledge of their molecular structure is more easily accessible with respect to their polymeric counterparts, allowing to ascertain precise structure–activity relationships, which can then be extrapolated towards those expected for polymers. Moreover, conjugated oligomers may present the same electrical and optical features of the related polydisperse high molecular weight materials.33
The versatility of the vinylsilanes arylation reaction described above was also exploited in a new methodology for the synthesis of well defined PPV oligomers (Scheme 12).34 The bis(α-trimethylsilylvinyl)benzene derivative 17, easily obtained by a double cross-coupling reaction of the aromatic Grignard reagent 20 with (bromovinyl)trimethylsilane 21, was converted into the dibromo bis(styryl) derivative 23 according to the Kikukawa protocol,30 in a Heck-type aryldesilylation reaction with the arenediazonium fluoroborate 22. This process was regio- and stereo-selective with respect to 22, affording 23 with all-E configuration. Moreover, the arylation reaction involved chemoselectively only the diazonium group of 22.
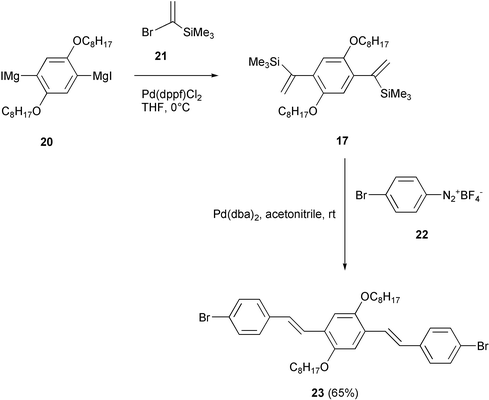 | ||
| Scheme 12 | ||
The extension of the conjugated system of 23 was performed by a formal coupling reaction with trimethysilyl derivatives 24, 25 and 26 (Scheme 13). The low reactivity of the simple trimethylsilyl unsaturated compounds in the transition metal-catalysed cross-coupling reactions was overcome by converting them into boron derivatives by reaction with boron trichloride,35 a procedure which represents a novel formal Suzuki–Miyaura type cross-coupling reaction starting with vinyltrimethylsilanes rather than boron derivatives. The oligomers 27a–c with 2n + 4 repeat units were obtained. The presence of at least two alkoxy groups is essential to allow solubility in organic solvents.
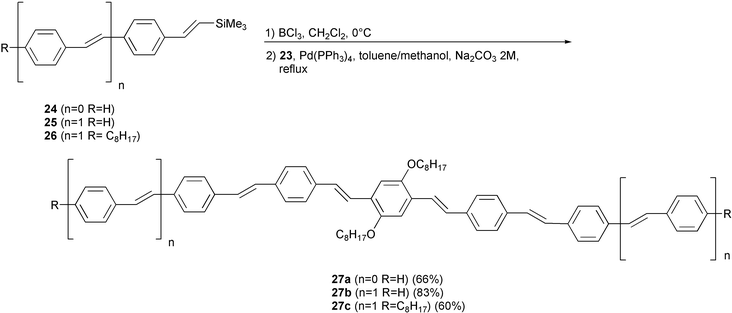 | ||
| Scheme 13 | ||
3. Arylation of alkynes (the Cassar–Heck–Sonogashira reaction)
The most frequently used synthetic tool for C–C bond formation between an sp and an sp2 carbon is represented by the Pd-catalyzed coupling of terminal alkynes with aromatic halides (bromides or iodides).36 Therefore, this process represents the preferred methodology for the synthesis of poly(phenyleneethynylene)s (PPE) and, more generally, poly(aryleneethynylene)s (PAE).37 The standard experimental conditions require the use of an amine as base, triethylamine, piperidine, morpholine, or pyrrolidine being the most used, and a copper(I) salt as cocatalyst, whose role is probably to activate the alkyne towards the transmetalation step by forming copper σ or π-acetylides.Although poly(1,4-phenyleneethynylene)s with alkyl or alkoxy susbstituents have been recently prepared by coupling suitable 1,4-diiodobenzenes with acetylene,38 bis(ethynyl) aromatic derivatives and dibromo- or more reactive diiodo-arenes, if easily available, are the preferred monomeric partners in this polymerization methodology. Electron-withdrawing substituents on the halide monomer improve the rate and yield of the coupling process. A very large variety of structures with an aromatic or heteroaromatic ring alternated with a C–C triple bond have thus been prepared by using almost the same experimental conditions, i.e. Pd(PPh3)4 or Pd(PPh3)2Cl2 as the catalyst, triethylamine as the base, and CuI, in toluene or THF. The formation of butadiyne defects is the major drawback of this polymerisation methodology. If Pd(II) complexes are employed, such defects are originated partly by the reduction of Pd(II) to Pd(0) in the catalyst activation step. However, the use of Pd(0) complexes, does not eliminate the occurrence of butadiyne units in the polymeric chain, deriving also from oxidative coupling of terminal triple bonds, which cannot be avoided even under strict exclusion of air. The source of the oxidizing agent remains unclear.
Examples of structures of substituted PPE with special interest in relation to the properties of the resulting materials, drawn from the recent literature, are reported in the following Scheme 14.
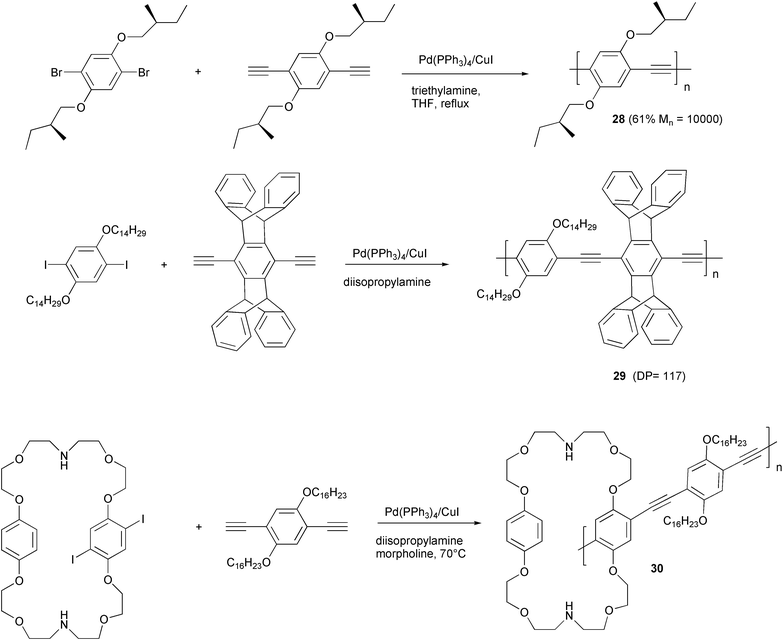 | ||
| Scheme 14 | ||
In PPE 28 the presence of chiral non-racemic alkoxy chains allowed the study of chiroptical properties in various solvents and in film.39 The comparison of CD spectra of film and solution in poor solvents such as alcohols or alcohol/chloroform mixtures showed unambiguously the strong aggregation of polymeric chains in solution.
In contrast, in the structure of PPE 29, obtained starting from diethynylpentiptycene, the presence of an extremely bulky structure prevents aggregation in the solid state, and films of this polymer are highly fluorescent owing to the reduced interchain interactions.40 Moreover, thin films of the same polymer may be used as an active layer for the fabrication of sensors for the detection of dinitro- and trinitro-toluene. Indeed, even low amounts of both aromatics are able to suppress reversibly the strong fluorescence of the polymer.
PPE 30,41 obtained by polymerisation reaction of the aromatic diiodo derivative with an azacrown structure and a dialkoxydiethynylbenzene, showed an unusual solid-state aggregation responsible for the highly emissive properties of the material. Films prepared by spin-casting or the Langmuir–Blodgett technique showed a different degree of aggregation of the material. Annealing or introduction of specific analyte (e.g. CO2) are able to induce de-aggregation processes. The consequent decrease in fluorescence may have potential applications in sensor technologies.
A specific functionalization at the end of polymeric chains of PPEs and related oligomers was made possible by the use of monomers bearing halogen atom and ethynyl function on the same aromatic ring. Donor- and acceptor-substituted oligo(phenyleneethynylene)s 31 were prepared as outlined in Scheme 15.42 An arene-palladium species, generated from iodonitrobenzene using the palladium catalyst, reacted with n equivalents of the AB monomer. The intermediate Pd-functionalized oligomer or polymer is end-capped by addition of aminoethynylbenzene. The synthesis of a PPE with thiols (protected as N,N-dimethylcarbamoyl derivative) end groups 32, was performed using N,N-dimethylcarbamoyl protected 4-iodothiophenol to generate the palladium intermediate and 4-ethynylthiophenol as the end capping reagent. Owing to their “molecular wire” nature, and to the presence of sulfur functionalities, these polymers can act as “molecular alligator clips” and bridge the gap between two gold nanoelectrodes.
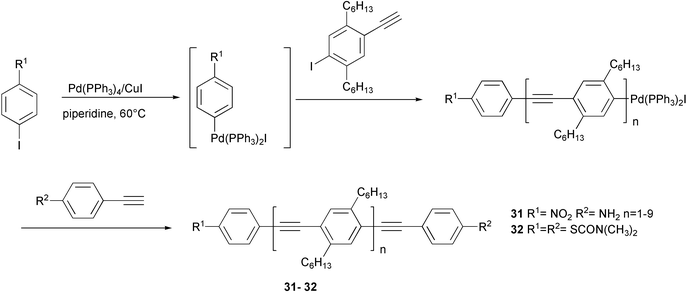 | ||
| Scheme 15 | ||
A polymer with one end of the polymeric chain capped with a norbornene ring 34 was prepared using a dialkoxy AB type monomer 33 and following a similar methodology (Scheme 16).43 The PPE chains were anchored on a silicon surface by means of the ring opening metathesis polymerisation (ROMP) with Grubbs catalyst. PPE “brushes” 35 were produced on the silicon substrate, which are structurally comb copolymers with PPE segments as pendant groups onto polynorbornene chains. The photoluminescence efficiencies measured for these films were higher if compared with the values measured for simple spin-cast films of the same PPE. As reported above, the aggregate formation generally leads to a quenching of photoluminescence due to the strong interchain interactions. In the case of the particular kind of film shown in Scheme 16, the enhanced emission efficiency was explained by supposing that single PPE chains are electronically isolated but strongly planarized in the brush structure. This observation opens new perspectives in fluorescent sensor applications.
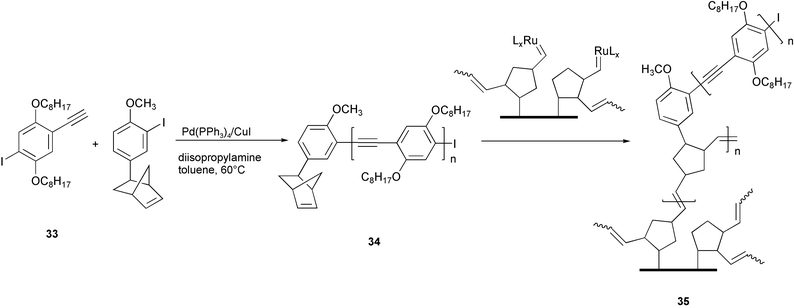 | ||
| Scheme 16 | ||
In the last few years, with the aim of creating organic semiconductors suitable for the realization of specific sensors for biological analytes, an increasing interest has grown around the synthesis of conjugated polymers functionalised with biomolecules.44 Although polythiophenes represent the most exploited class for these applications, some examples of PPE bearing monosaccharide units as substituents have been recently reported. The synthesis of various “sugar coated molecular wires” 36 (Scheme 17)45 was performed adopting a novel palladium-catalyzed coupling of trimethylsilyl alkynes with aromatic halides using silver oxide to replace both copper cocatalyst and amine.46 Transesterification with catalytic sodium methoxide in methanol allowed an easy deprotection of the glucose moieties, and the resulting material showed an enhanced solubility in polar solvents.
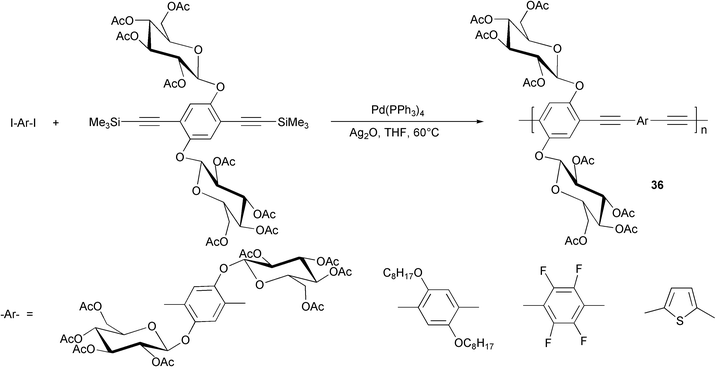 | ||
| Scheme 17 | ||
The comparison of spectral data of these polymers and those of 37, which was prepared by the same methodology starting from 1,4-bis(trimethylsilyl)butadiyne (Scheme 18) allowed the absence of diyne defects to be ascertained.
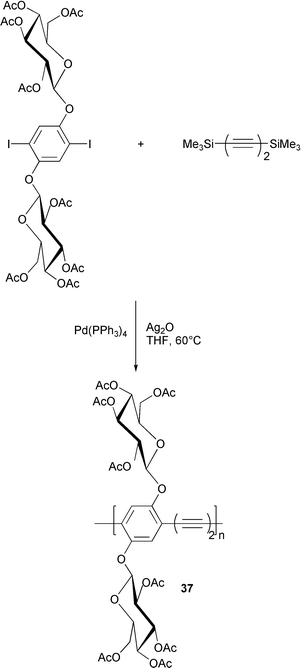 | ||
| Scheme 18 | ||
Similar PAEs47 have also been prepared by reacting a penta-O-acetylglucose functionalized aromatic dihalide with a 2,5-dialkyl-1,4-bis(ethynyl)benzene in the standard Cassar–Heck–Sonogashira coupling conditions.
4. Cross-coupling reaction of boron derivatives (the Suzuki–Miyaura coupling)
Oligo- and poly-(phenylenevinylene)s, -phenylenes, -(phenyleneethynylene)s and -thiophenes can be conveniently prepared by means of the Pd-catalyzed cross-coupling reaction of an aromatic diboron derivative with a vinyl- or aryl-dihalide (or triflate), in the presence of a base (the Suzuki–Miyaura cross-coupling reaction48). The methodology is able to provide these classes of conjugated polymers with high structural variety, owing to the compatibility of the reaction conditions with a wide range of functional groups. However, a careful choice of reaction parameters, such as the palladium complex ligand, the base/solvent system, and the nature of the boron derivative, is required in order to achieve high molecular weights and good yields. Optimal coupling conditions may vary with individual monomers and often good results are obtained only after an extended "trial and error" investigation. The synthesis of alkyl-susbstituted oligo(p-phenylenes) 38 is an example of the application of this coupling reaction starting with aromatic dihalides and aromatic diboronic acids, which are the most used boron derivatives, in the standard reaction conditions (Scheme 19).49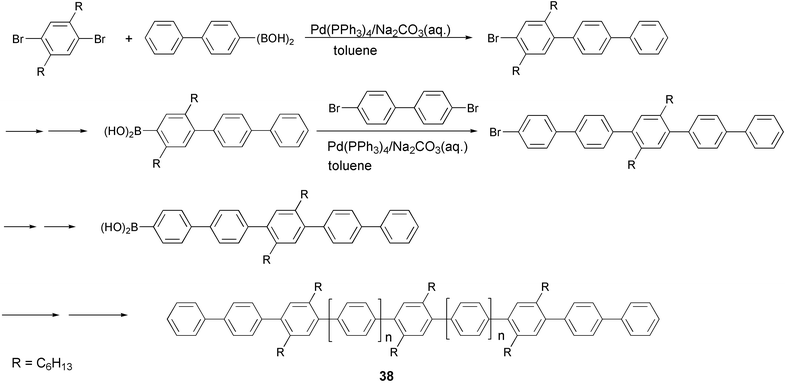 | ||
| Scheme 19 | ||
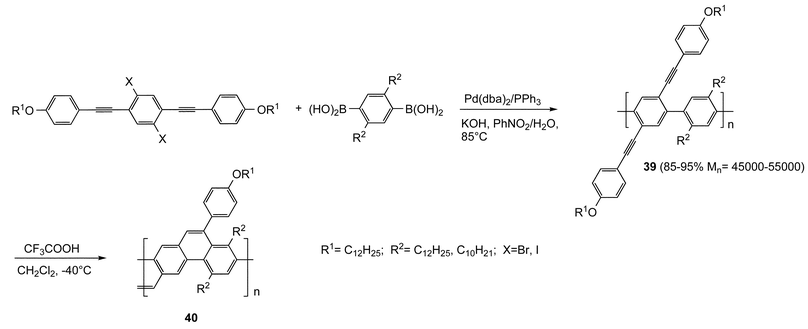
In the strategy adopted for building the main chain of the PPP 39 (Scheme 20)50in situ generation of the active catalytic species from Pd(II) complex with a labile ligand, such as Pd(dba)2 (dba, dibenzylideneacetone), and a suitable amount of triphenylphosphine is preferred as a useful alternative to the use of air-sensitive Pd(0) phosphine complexes. The electrophile-induced cyclization reaction with trifluoroacetic acid converts the linear polymer in a ladder PPP structure 40 which, due to its planarity, allows a maximized π-conjugation extension.
 | ||
| Scheme 20 | ||
Although aromatic boronic acids are widely employed in the Suzuki–Miyaura coupling owing to their easy preparation and air-stability, their purification often is not easy. In many cases, cyclic boronic esters, which can be obtained with higher purity from the corresponding acids, are conveniently used.
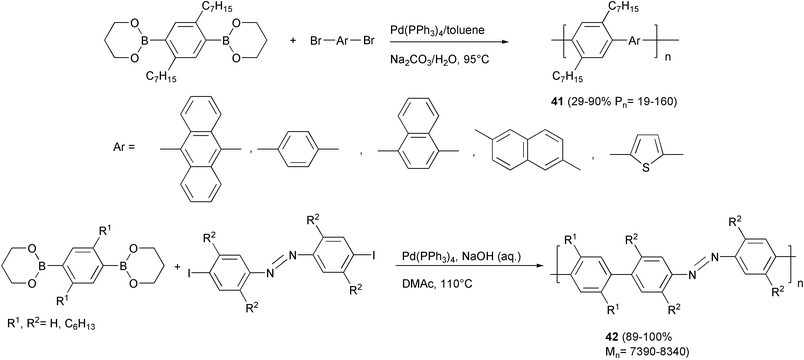
Examples of the use of propanediol boronic esters are represented by the synthesis of various polyarylenes 41 (Scheme 21) with strong photoluminescence in the blue region,51 and of PPP polymers with azobenzene units in the main chain 42.52 The presence of photoisomerizable azobenzene units makes this new class of conjugated polymers particularly promising in the field of photoresponsive materials, whose electrical and optical properties can be modified upon photoirradiation at the appropriate wavelength.
 | ||
| Scheme 21 | ||
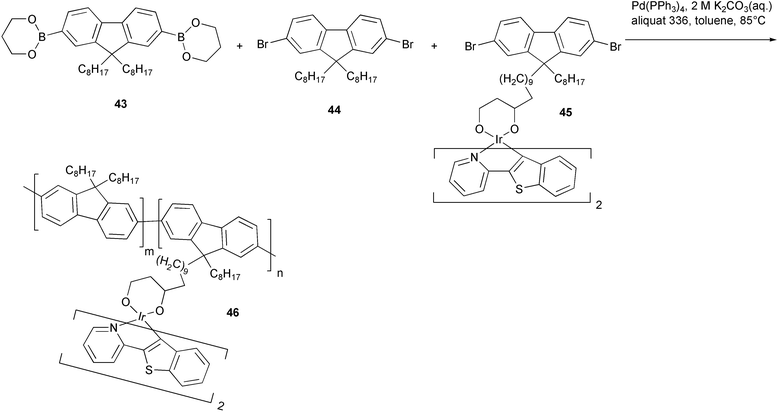
Red-emitting electroluminescent devices have been recently fabricated using polyfluorenes grafted with a cyclometalated red phosphorescent iridium complex 46 (Scheme 22).53 These materials were obtained by Suzuki–Miyaura copolymerisation, performed in the presence of a phase transfer agent (aliquat 336), of the fluorene propanediol boronic ester 43 with different feed ratios of the fluorene dibromides 44 and 45 having the iridium complex as pendant group. Phenyl propanediol boronic ester and tert-butylbromobenzene were added sequentially as capping agents at the end of the polymerization reaction. Energy transfer from the blue emitting polyfluorene chain to the phosphorescent iridium complex occurred with high efficiency, improving the electroluminescence efficiency of the devices.
 | ||
| Scheme 22 | ||
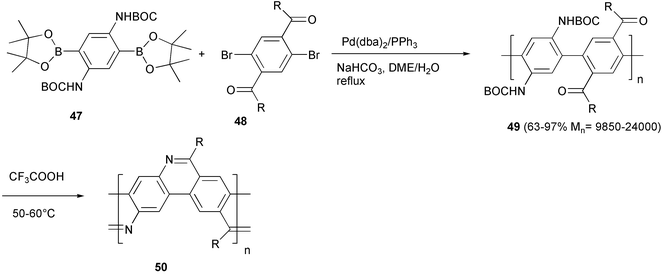
Pinacol borate esters are characterized by a higher stability to hydrolysis than the corresponding 1,3-propanediol derivatives, and are easily prepared by direct reaction of pinacol with dimethyl boronic esters. Moreover, they can be purified by conventional chromatographic techniques. The synthesis of the imine bridged planar poly(p-phenylene) derivative 50 was performed using the pinacol borate ester 47, which was coupled with the dibromoderivative 48 using a Pd(0) complex generated in situ from Pd(dba)2/PPh3, and NaHCO3 in 1,2-dimethoxyethane/water (Scheme 23).54 The linear PPP polymer 49 is converted into the ladder structure by imine formation between the carbonyl and amino groups of adjacent rings.
 | ||
| Scheme 23 | ||
In spite of the high synthetic potential of this organometallic strategy for the synthesis of polyconjugated materials, problems are faced with some side reactions which occur in the coupling process and have only a limited impact on the synthesis of small molecules, but become of significant importance in the polymerization because they may lead to end-caps and to the presence of structural defects in the polymeric chains. A detailed study performed by Novak and coworkers on the synthesis of PPP polymers by Suzuki–Miyaura coupling evidenced the formation of branched isotropic materials instead of a linear anisotropic rod-like structure.55 Such structural defects derive from the aryl exchange reaction between one or more phenyl rings of the phosphine ligand and the palladium-bound growing polymeric chain in the catalytic cycle and generate phenyl end-caps and structural defects including the phosphorus atom (Scheme 24).
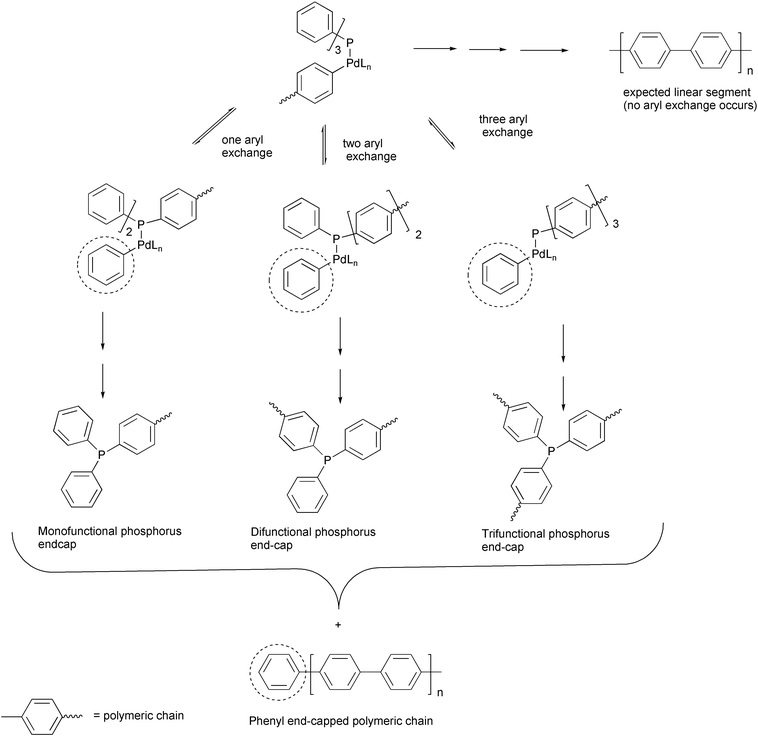 | ||
| Scheme 24 | ||
31P NMR spectra of polymer 51 (Scheme 25), prepared using Pd(PPh3)4 as conventional catalyst in THF/water and potassium carbonate as base, revealed the presence of a phosphorus signal in the phosphine oxide region (δ = 28.5 ppm), indicating that a substantial amount of phosphine was incorporated in the polymer backbone. This side reaction can be strongly reduced by choosing the bulky tri(o-tolyl)phosphine (TOTP) as palladium ligand. Polymers obtained using Pd(TOTP)4 are of higher molecular weight and 31P NMR spectra show the absence of any phosphorus signal.55
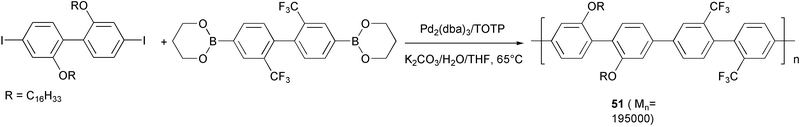 | ||
| Scheme 25 | ||
A similar detailed study on the influence of side reactions occurring in the Suzuki–Miyaura coupling was performed on the polymerization reaction of thiophene 2,5-diboron derivatives 52–54 with 1,4-diodo-2,5-dialkoxybenzenes 55 and 56 to give poly(arylenethienylene)s 57 and 58 (Scheme 26).56 In this case the effects on molecular weights, yields, polydispersity, and end groups of the polymers obtained either by using Pd(PPh3)4 or phosphine-free Pd(OAc)2 catalysts were investigated in THF/water and potassium carbonate as base, in relation to the thiophene boronic derivative used as comonomer. As a general result, the use of Pd(OAc)2 had a marked positive effect on the molecular weights of the copolymer with respect to the phosphine palladium complex, as a consequence of the phosphine inhibition that may limit the catalytic efficiency in the Suzuki–Miyaura aryl coupling.57 The nature of the boron derivative may also influence markedly the molecular weight of the polymers. The pinacol diester 54 gave better results in terms of molecular weights with respect to the related 1,3-propanediol ester 53 or 2,3-thiophene diboronic acid 52.
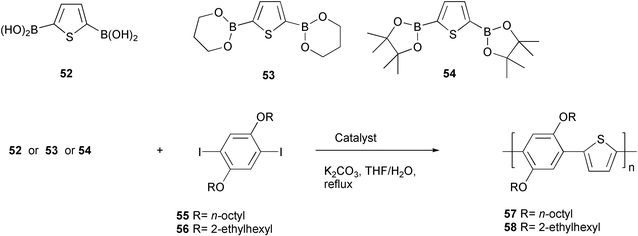 | ||
| Scheme 26 | ||
The phosphine-free catalyst Pd(OAc)2 was also employed in the synthesis of regioregular alkyl-substituted oligothiophenes, and, in particular, sexithiophenes. These materials are the most used organic semiconductors for the fabrication of field effect transistors (FET), owing to their self-assembling properties which make it possible to obtain high molecular order in thin films, an essential prerequisite for high mobilities of charge carriers. Unsubstituted sexithiophene appears as a very promising material for FET fabrication, but its low solubility does not allow it to be obtained in a pure form. The presence of alkyl chains improves the solubility and makes the purification easier. However, in order to preserve the good self-assembly properties of the material, a regular disposition of alkyl chains in the oligomer structure is required.
Soluble regioregular sexithiophene 59 was obtained very efficiently by microwave assisted Suzuki–Miyaura cross-coupling in only 5 minutes at 25 W (Scheme 27).58 The same coupling performed under typical Suzuki–Miyaura conditions did not allow the oligomer to be obtained in acceptable yield. The same product could be prepared, in much higher yield, in a solvent-free microwave-assisted process using PdCl2(dppf) on alumina with KF as base, which allowed a more simple purification of the reaction product by filtration.59
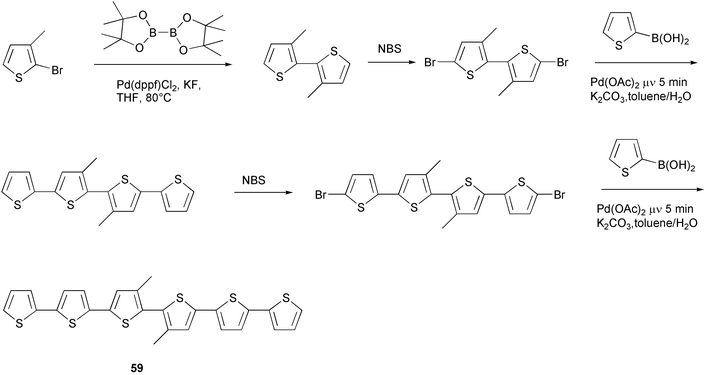 | ||
| Scheme 27 | ||
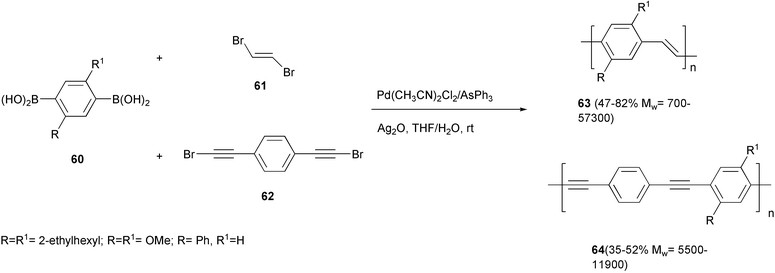
The Suzuki–Miyaura coupling has found a lower number of applications in the synthesis of PPV and PPE. PPV 63 and the PPE 64 were obtained by the coupling reaction of trans-1,2-dibromoethylene 61 or 1,4-bis(2-bromoethynyl)benzene 62, respectively, with alkoxy- or phenyl-substituted 1,4-benzenediboronic acids 60 using Pd(AsPh3)4 generated in situ, with silver oxide as base in THF/water (Scheme 28).60
 | ||
| Scheme 28 | ||
An accelerating effect on the rate of the reaction was observed when silver oxide was used as base. The coupling reactions could be performed even at room temperature. Low amounts of homo-coupling product of the boronic acid were detected in the model reactions of benzeneboronic acid with dibromoethylene or (2-bromoethynyl)benzene under the same conditions. Indeed, 13C NMR spectra evidenced the presence in the polymeric materials of ca. 3% of biphenyl units, derived from the homo-coupling reaction.
5. Cross-coupling reaction of tin derivatives (the Stille coupling)
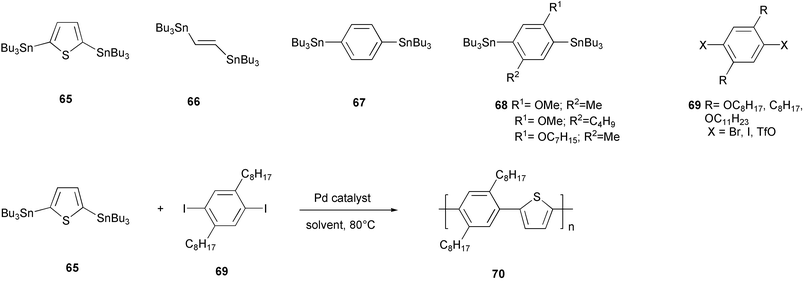
The use of aryl or vinyl organostannanes in Pd-catalyzed coupling with aryl halides or triflates61 represents an alternative methodology to the Suzuki–Miyaura coupling in building C–C bonds in well defined molecules, and the extension of this process to the synthesis of polymeric conjugated materials was also widely investigated.A detailed study on the Stille coupling possibilities was performed by Yu and coworkers, by considering the polymerization reaction of stannanes 65–68 with the diiodo aromatic derivatives 69 bearing alkyl or alkoxy substituents (Scheme 29).62 The effects of solvent and catalyst were investigated in the polymerization of 2,5-bis(tributylstannyl)thiophene 65 with the diiododerivative 69 (R = C8H17, X = I). In THF as solvent, Pd(AsPh3)4, generated in situ by mixing the weakly coordinated Pd2(dba)3 with four equivalents of triphenylarsine, showed the higher stability and catalytic efficiency, allowing the synthesis of relatively high molecular weight polymers.
 | ||
| Scheme 29 | ||
Moreover, the phosphine-free ligand prevented the aryl-aryl exchange in the active palladium catalytic species, which may lead, as previously discussed for the Suzuki–Miyaura coupling, to a low molecular weight polymer, due to the endcap with a phenyl ring of the polymeric chain.
The extension of this study to the polymerization of the tin monomers 66–68 evidenced the higher reactivity of the thiophene distannylated monomers with respect to the other tin derivatives, and the Stille coupling appeared as a useful polymerization process mainly for preparing thienylene type polymers, by using the reactive 2,5-bis(tributylstannyl) derivatives and, indeed, a great number of this kind of conjugated polymers have been prepared by Stille polymerization.
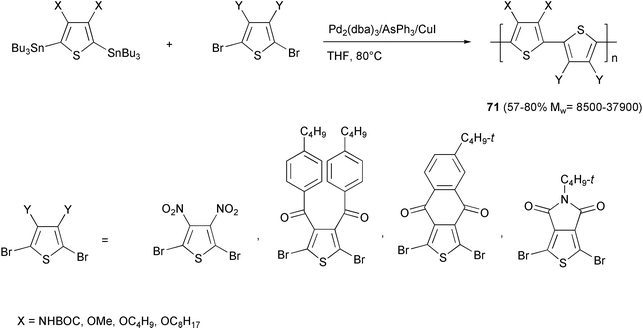
A series of polythiophenes with alternation of donor/acceptor units 71 (Scheme 30)63 were obtained adopting the optimized experimental conditions reported (i.e.in situ generated Pd(AsPh3)4 in THF). The addition of CuI64 was found to have a positive effect on the molecular weights. The regular alternation of electron-rich and electron-deficient repeat units induces a high planarity of the conjugated backbone by intramolecular charge transfer, producing a low band gap polythiophene polymer.
 | ||
| Scheme 30 | ||
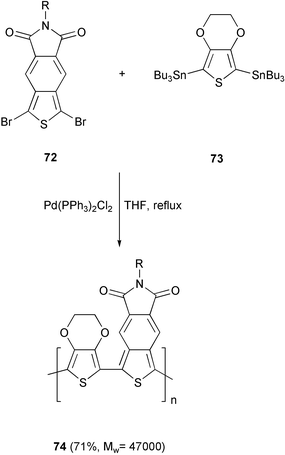
Other low-bandgap polythiophenes are represented by the copolymer 74 obtained by a Stille cross-coupling reaction between the 2,5-bis(tributylstannyl)-3,4-ethylenedioxythiophene 73 with the thiophene derivative 72 (Scheme 31).65 Thin films of this material are transparent in the visible region, but show an electrochromic behaviour at 1500 nm, which may be useful for specific infrared device application.
 | ||
| Scheme 31 | ||
Although the studies on the Stille polymerisation have pointed out the low reactivity of the 1,2-bis(tributylstannyl)ethene 66, this compound was employed in the synthesis of arylenevinylene type polymers. Polythienylenevinylene 76 was obtained in relatively high molecular weight by the cross-coupling reaction of 66 with diiodothiophene 75 (Scheme 32).66
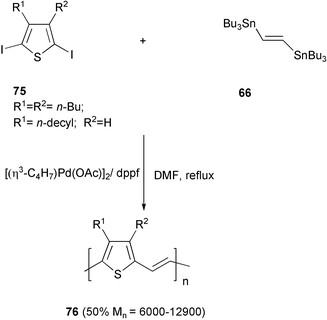 | ||
| Scheme 32 | ||
Moreover, several poly(p-phenylenevinylene)s with linear, branched or bridged alkoxy, polyethereal or silyloxy chains were successfully prepared by coupling the same organotin monomer 66, in benzene with Pd(PPh3)4 as catalyst, with various diiodoarenes 77 (Scheme 33).67 These polymers showed a good solubility in organic solvents, which allowed the preparation of quality films by spin-coating techniques, despite the limited values of the molecular weights. Attractive optical properties, such as stimulated emission in solution and thin film, were observed for PPVs 79 and 80. PPV 80 with bridged alkoxy chain showed an improved solid state photoluminescence efficiency with respect to the open chain polymers 78 and 79. The presence of the bulky cyclic chain onto the aromatic ring is responsible for the reduction of interchain interactions, which increase the emission efficiency.68 The interesting properties observed, related also to the high regularity of the conjugated structure, make these materials an interesting alternative to alkoxy PPVs prepared by Wessling or Gilch polymerisation methodologies which, as pointed out in the Introduction, may present structural and conjugational defects.
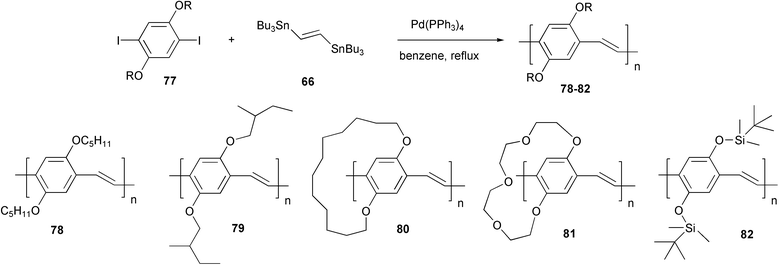 | ||
| Scheme 33 | ||
An example of particular interest is represented by PPV 84, the first PPV with all benzene rings substituted with fluorine atoms, obtained by modified Stille cross-coupling conditions, using Pd(AsPh3)4 generated in situ from Pd2(dba)3/AsPh3 in benzene (Scheme 34).69 The replacement of C–H bonds with stronger and less reactive C–F bonds may have a positive effect on the resistance against degradation phenomena and is expected to shift the emission wavelength towards the blue region of the visible spectrum. This effect was actually detected in an electroluminescent device, fabricated with a thin film of this material obtained by thermal evaporation. The emission wavelength was shifted in the green region of the visible spectrum, with respect to the red–orange emission of alkoxy-substituted PPVs.
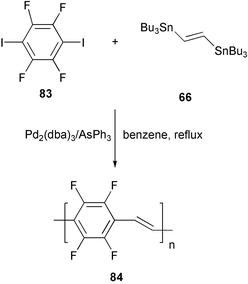 | ||
| Scheme 34 | ||
The Stille cross-coupling reaction was also exploited for the synthesis of copoly(2,3,5,6-tetrafluoro-1,4-phenylenevinylene-2,5-dialkoxy-1,4-phenylenevinylene)s 85 containing variable ratios of dialkoxyphenylene and tetrafluorophenylene monomeric units (Scheme 35).70 The great polarizability of the conjugated system of these materials, originated by the presence of phenylene rings substituted with either electron-donor (alkoxy) or electron-withdrawing (fluorine) side groups, exerts a marked influence on the third-order NLO properties. In particular, the χ(3) values measured in chloroform solutions of these different copolymers are up to one order of magnitude larger than the value detected for the corresponding dialkoxy-substituted homo-polymer.
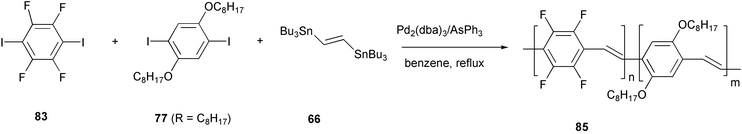 | ||
| Scheme 35 | ||
6. Transition metal catalyzed cross-coupling reactions with organomagnesium or organozinc reagents
Polyphenylenes and polythiophenes have also been prepared by means of cross-coupling reactions of organomagnesium (the Kumada–Corriu coupling)71 or organozinc (the Negishi coupling)71 reagents with aryl halides in the presence of palladium or nickel complexes as catalysts.The polymerization of 1,4-dibromobenzene in the presence of magnesium and NiCl2(bpy) (bpy, bipyridine) to afford insoluble poly(1,4-phenylene) was reported by Yamamoto and coworkers.72 Soluble low molecular weight (6–7 units DP) poly(2,5-dialkyl-1,4-phenylene)s were obtained in a subsequent work, performed by Schlüter, Feast and coworkers, starting from dialkyl-substituted dibromobenzenes.73 The growth of polymeric chains may be prevented by side reactions, such as partial reduction of bromine end groups, or hydrolysis of the Grignard species by adventitious water. In these processes a bromomagnesiobromobenzene is assumed as intermediate species, which then undergoes a self-coupling reaction in the presence of the Ni(II) catalyst.
In the synthesis of polythiophene developed by Heeger, Wudl and coworkers, the iodomagnesioiodothiophene intermediate deriving from highly purified 2,5-diiodothiophene with magnesium in diethyl ether was isolated as a residue, and the polymerization reaction was performed in anisole in the presence of Ni(dppe)Cl2 as catalyst. A relatively high molecular weight polythiophene was obtained (Mn = 4000).74
As the most significant result in the application of this organometallic strategy to the synthesis of conjugated polymers, it is worth mentioning that regioregular head-to-tail (HT) poly(3-alkyl-2,5-thiophene)s (PATs) were obtained following two different methodologies based upon the use of magnesium- or zinc-thiophene intermediates.
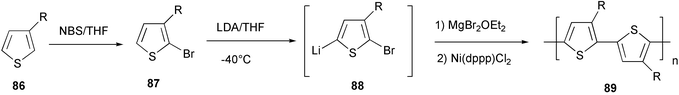
In the magnesium route (the McCullough methodology), bromination of 3-alkylthiophene 86 occurred selectively at the 2-position, affording 2-bromo-3-alkylthiophene 87 (Scheme 36).75
 | ||
| Scheme 36 | ||
Regioselective metalation of this thiophene halide with lithium diisopropylamide at the 5-position gave origin to a bromo lithium thiophene intermediate 88, which is stable at low temperature and does not undergo metal halogen exchange. The conversion of 88 into the corresponding magnesium derivative by exchange with MgBr2·Et2O was followed by the polymerization step to 89, in the presence of a Ni(II) complex.
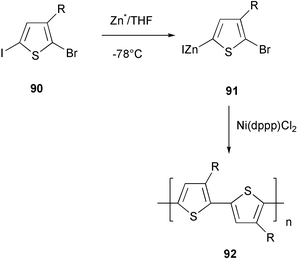
The 3-alkyl-2-bromo-5-(iodozincio)thiophene 91, which was polymerized in the presence of a Ni(II) catalyst, is the key intermediate in the zinc strategy. This organometallic species is generated regioselectively from 3-alkyl-2-bromo-5-iodothiophene 90 by using the active “Rieke zinc” (Scheme 37).76
 | ||
| Scheme 37 | ||
The choice of the metal complex as catalyst is of relevance in both methodologies, in relation to the homo-coupling of the substrate, which is the side reaction in the polymerization step. It was observed that Ni complexes with bidentate ligand gave exclusively cross-coupled products affording highly regular HT PATs, whereas with Pd complexes bearing monodentate ligand a random mixture of cross-coupled and homo-coupled products was produced. Ni(dppp)Cl2 was the catalyst which afforded the best results in terms of structure regularity.
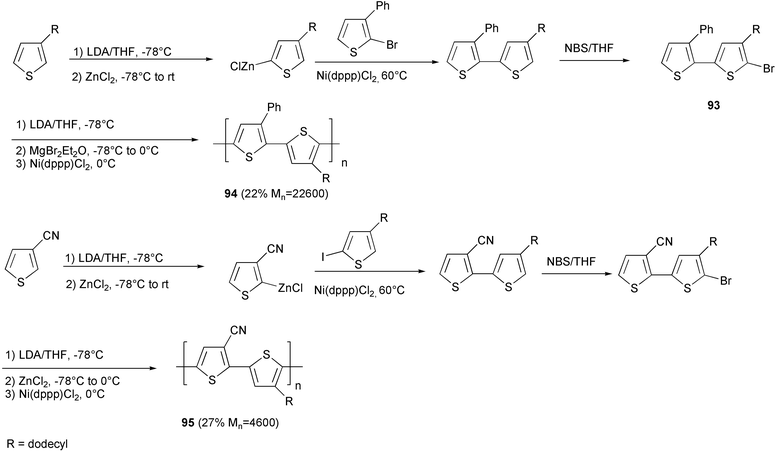
The synthesis of HT polythiophene with regular alternation of phenyl-substituted and alkyl-substituted units 94 was performed by the McCullough methodology starting with the suitable bisthiophene monomer 93 (Scheme 38).77 A modification of this method by converting the lithium intermediate into the zinc derivative instead of the magnesium derivative was employed in the preparation of the polythiophene 95, presenting the alternation of cyano- and alkyl-substituted units.
 | ||
| Scheme 38 | ||
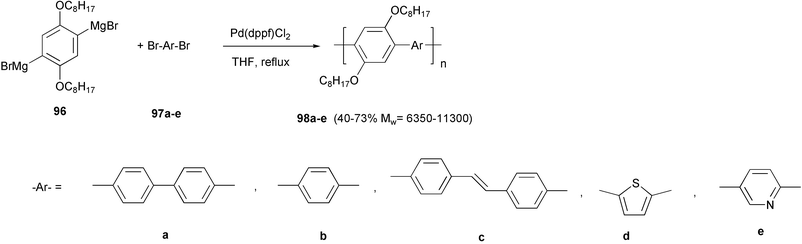
A series of polyarylene and poly(arylenevinylene) type polymers 98 was obtained by the cross-coupling reaction of the dibromomagnesium derivative 96 with the appropriate dihaloarenes 97 (Scheme 39).78 The polymerization of 96 with 97a was chosen as the model system, and the best reaction conditions (THF as solvent, rt) and the most appropriate catalyst [Pd(dppf)Cl2] were selected in order to obtain the highest yields and molecular weights. MALDI-TOF analysis of terminal groups revealed in these polymers the presence of halogen atoms at one or both ends of the polymeric chains. Therefore, the reductive dehalogenation reaction appeared to play a minor role in terminating the polymeric chains, and the precipitation of the growing chains was considered the main factor responsible for the prevention of their further growth. A Langmuir–Schäfer film of the thienylenearylene polymer 98d was employed as active layer in the fabrication of a resistive chemical gas sensor with excellent sensitivity towards NO2, reversibility, and time stability of the response.79
 | ||
| Scheme 39 | ||
7. Conjugated materials by homo-coupling reactions
Polyphenylenes, polythiophenes, and thiophenephenylene random copolymers 100 were obtained by the homo-coupling reaction of aromatic halides 99 promoted by Ni(0) complexes (Scheme 40).80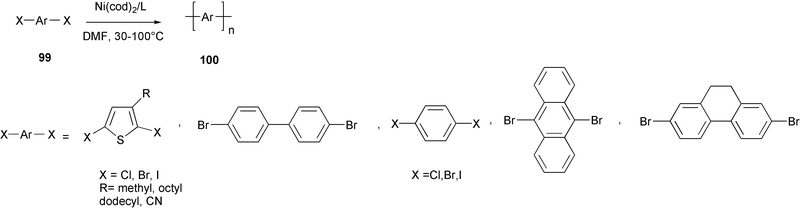 | ||
| Scheme 40 | ||
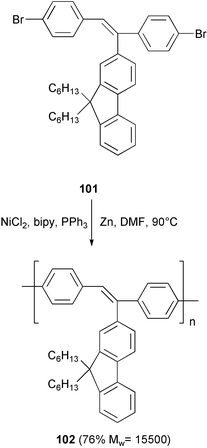
The blue-electroluminescent polymer 102, with a poly(biphenylenevinylene) structure bearing bulky fluorenyl groups as substituents on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds, was prepared by homo-coupling of the 4,4′-dibromostilbene derivative 101 promoted by Ni(0) generated in situ by reduction of NiCl2 with zinc in the presence of triphenylphosphine and bipyridine (Scheme 41).81
C double bonds, was prepared by homo-coupling of the 4,4′-dibromostilbene derivative 101 promoted by Ni(0) generated in situ by reduction of NiCl2 with zinc in the presence of triphenylphosphine and bipyridine (Scheme 41).81
 | ||
| Scheme 41 | ||
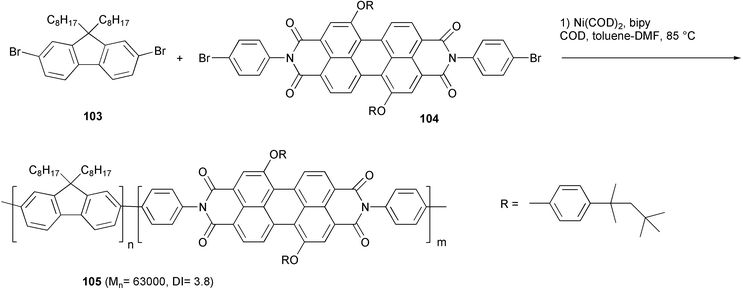
Fluorene based polymers are materials with electroluminescence in the blue region, but their emission color can be tuned by incorporating a perylene dye in the main chain.82 Such a type of copolymer is exemplified by the structure 105 (Scheme 42), which was obtained from Ni-catalyzed homo-coupling of the dibromofluorene 103 with the dihaloperylene dye 104. The emission originated solely from the dye units, owing to an efficient energy transfer from the polyfluorene segments to the dye chromophore.
 | ||
| Scheme 42 | ||
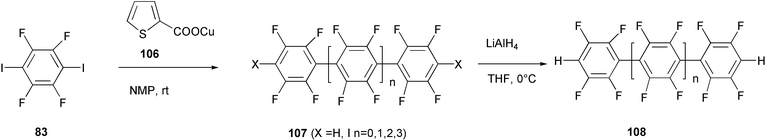
The Ullmann-like reductive coupling of aryl halides was employed for the synthesis of fluorinated PPP oligomers 108 starting from the diiodo tetrafluoro derivative 83 (Scheme 43).83 The air stable copper(I) thiophene carboxylate 106 was the specific promoter of this homo-coupling reaction in N-methylpyrrolidinone as solvent. A complex mixture of oligomers 107 containing up to five aromatic rings was obtained. A quantitative dehalogenation reaction with lithium aluminium hydride in THF led to the four fluorinated PPPs 108 with hydrogen atoms as terminal groups, which could be separated by flash chromatography. The introduction of fluorine atoms as substituents on the aromatic rings should increase the HOMO–LUMO energy gap, making these materials potentially appropriate for blue-emitting devices. Moreover, a greater chemical stability is expected by replacing C–H bonds with less reactive C–F bonds.
 | ||
| Scheme 43 | ||
Conjugated polyenes represent an interesting class of oligomers acting as a discrete model of polyacetylene. Moreover, they are well-known to exhibit non-linear optical properties.
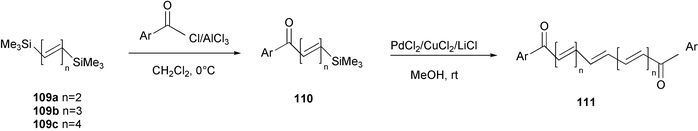
1,4-Bis(trimethylsilyl)-1,3-butadiene 109a, 1,6-bis(trimethylsilyl)-1,3,5-hexatriene 109b, and 1,8-bis(trimethylsilyl)-1,3,5,7-octatetraene 109c were used as convenient building blocks for the synthesis of α,ω-diketopolyenes containing up to eight all-E conjugated double bonds (Scheme 44).84 A stereoselective homo-coupling reaction of polyunsaturated silanes 110, promoted by the PdCl2/CuCl2/LiCl system in methanol, represented an easy synthetic route to the symmetrically substituted polyenes 111, which are materials with potentially large third-order non-linear optical coefficients. The silylated ketones 110 are easily available by a chemoselective acylation reaction of the polyunsaturated silanes 109.
 | ||
| Scheme 44 | ||
8. Metathesis reactions
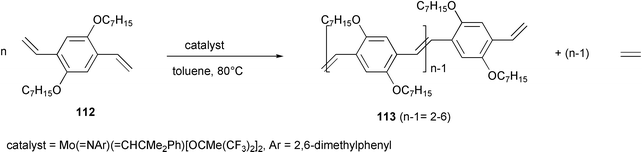
ADMET (acyclic diene metathesis) or ADIMET (acyclic diyne metathesis) processes were employed in the synthesis of double- or triple-bond containing conjugated polymers. Moreover, the ROMP (ring opening metathesis polymerization) process was exploited as a precursor polymer route to polyacetylenes and poly(phenylenevinylene)s.Dialkyl- or dialkoxy-substituted PPV oligomers with 3–7 repeat units 113 were prepared, following the ADMET methodology, by oligomerization of bis(styryl) benzene derivative 112 in the presence of a Schrock type molybdenum alkylidene complex (Scheme 45).85
 | ||
| Scheme 45 | ||
Monomers bearing alkoxy-substituents were much less reactive with respect to the alkyl-substituted ones, and the donor ligand action of the alkoxy oxygen towards the molybdenum atom, which stabilizes the intermediate alkylidene metallacyclobutane complex, was proposed as an explanation.
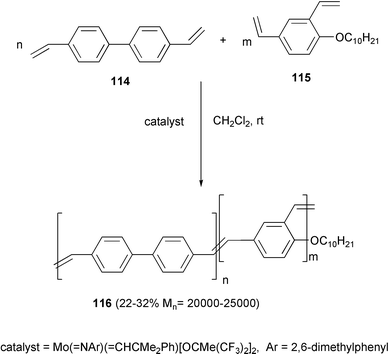
Poly[(m-phenylenevinylene)-co-(p-phenylenevinylene)] 116 with higher molecular weight was obtained by the same methodology, starting with the bis(styryl) derivatives 114 and 115 (Scheme 46).86 These polymers are characterized by a strong photoluminescence in thin films and a progressive blue-shift of the emission wavelength was evidenced in relation to the content of meta repeat units.
 | ||
| Scheme 46 | ||
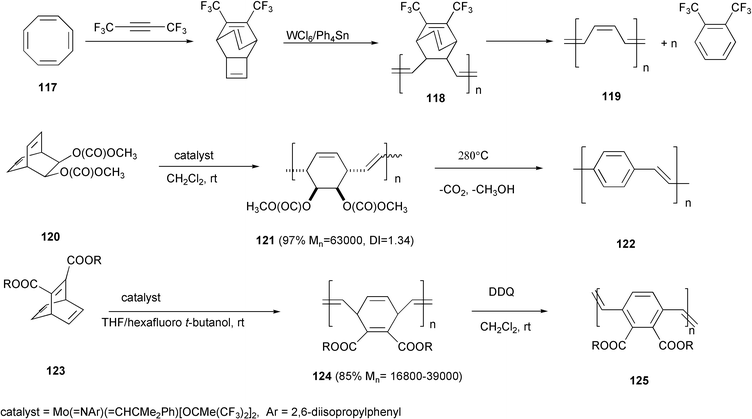
The ROMP methodology afforded polymer 118, non-conjugated precursor of polyacetylene 119, starting from cyclooctatetraene monomer 117.87 Similarly, polymers 121 and 124, precursors of poly(phenylenevinylene)s 122 and 125, respectively, were obtained from bicyclo[2.2.2]octadiene88120 or barrelenes89123 (Scheme 47). The conjugated polymers are generated by thermal elimination or chemical oxidation of these precursors. In some cases the stereochemistry of the double bonds originated in the metathesis process is not defined, and near equal amounts of cis- and trans-vinylene units are present. However, in the following step of elimination or oxidation a cis–trans isomerization occurs and an all trans stereochemistry of the conjugated vinylene units results. High molecular weight and near unity polydispersity of the polymers derive from the “living” characteristics of the polymerization process. Polyacetylenes were also prepared directly by ROMP starting from cyclooctatetraene derivatives.90
 | ||
| Scheme 47 | ||
PPE polymers were obtained by means of the ADIMET process using Schrock's tungsten-carbine complex (t-BuO)3WCt-Bu.91 However, molybdenum hexacarbonyl and 4-chlorophenol (the Mortreux catalyst92) in 1,2-dichlorobenzene at 150°C represented a useful alternative catalytic system, successfully employed in the polymerization or copolymerization of alkyl- or alkoxy-substituted 1,4-dipropynylbenzenes 126 to give the corresponding PPE polymers 127 in high yields and polymerization degrees. In some cases, further activation of the catalyst by 4-trifluoromethylphenol is needed (Scheme 48).93
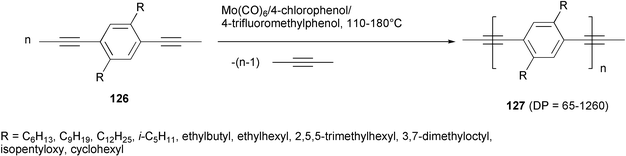 | ||
| Scheme 48 | ||
Many advantages are offered by this catalytic system with respect to the Schrock catalyst, which is rather difficult to prepare and is air- and moisture-sensitive. In contrast, these molybdenum-based catalysts are inexpensive, air stable and work in non-dried, non-purified off-the-shelf solvents. This methodology was also applied to the synthesis of PPVE polymer 129, a hybrid of the structure of PPE and PPV, starting from the dipropynyl stilbene 128 as monomer and taking advantage of the great tolerance of this metathesis catalytic system with respect to the double bond, which is not involved in the process94 (Scheme 49).
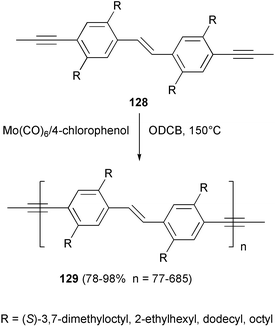 | ||
| Scheme 49 | ||
9. Transition metal catalyzed polymerization of alkynes to polyacetylenes
Polyacetylenes can be prepared from the corresponding acetylenic monomers by Ziegler–Natta type polymerization with MoCl6, WCl6 or TaCl5 based catalysts,95 or by using organorhodium(I) complexes.96A well-defined polyacetylene 131 with double bond moieties in the side chain was derived from enyne 130. The use of rhodium dinuclear complexes in triethylamine or THF as solvent allowed the regioselective polymerization involving only the triple bond of the monomer (Scheme 50).97
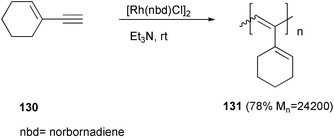 | ||
| Scheme 50 | ||
A rhodium complex, generated in situ starting from an organorhodium precursor [Rh(nbd)(OMe)]2 and an equivalent amount of a bidentate phosphine (i.e. dppe, dppp or dppb) allowed a stereoselective polymerization of phenylacetylene 132 (Scheme 51).98 Poly(phenylacetylene) 133 was obtained in the cis-transoid stereoregular form in high molecular weight and low polydispersity, thus suggesting the “living” nature of the polymerization, which was effectively proved by the increase of the polymer chain length upon adding further monomer after 100% conversion.
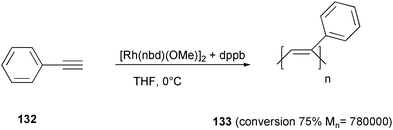 | ||
| Scheme 51 | ||
WCl6 or TaCl5 in the presence of Bu4Sn or Ph3Bi were used as the catalytic systems for the polymerization of acetylene derivatives 134, 135 or the butadiyne derivatives 138, 139.99 A novel class of polyacetylenes with pendant pyrenyl groups bound to the main chain, either directly or indirectly via an additional acetylene linkage, was obtained with high molecular weights, but with rather high polydispersities (PDI = 1.8–11) (Scheme 52). Polymers 136 and 140 showed a higher degree of conjugation than other polyacetylenes. Furthermore, interactions between the pyrene units in the chains are present, as revealed by emission bands in the photoluminescence spectra due to the associated pyrenes. This association appeared significantly weakened in the polymers 137 and 141, owing to the steric hindrance in the conjugated backbone originating from the presence of the trimethylsilyl group.
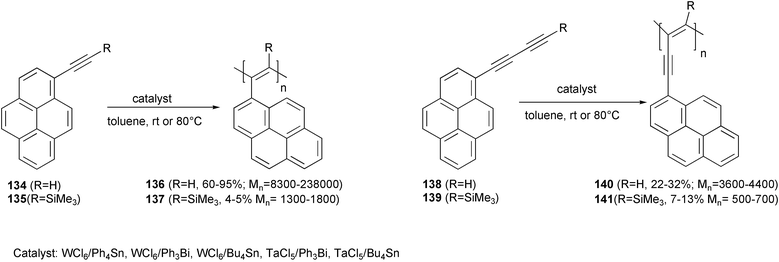 | ||
| Scheme 52 | ||
Polyacetylenes bearing mesogenic side groups are materials of great interest in the field of liquid crystals. The light emitting properties of some of these materials may also be exploited as a polarized light source in display technology. Moreover, temperature changes or application of an external electric or magnetic field may modify the relative orientation of the mesogenic side groups. As a consequence, conformational rearrangements of the conjugated backbone could derive, in which the main chain conjugation is improved by an enhanced coplanarity. When chiral mesogenic side-groups were present, as in polyacetylene 142 (Scheme 53),100 conformational changes were detected by CD measurements. A helical screw inversion most likely takes place in an extremely narrow temperature range, and this behavior may be exploited in constructing a two-state temperature-switchable device.
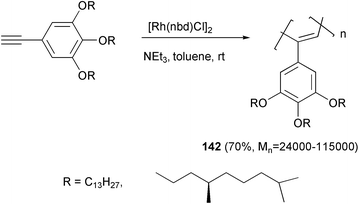 | ||
| Scheme 53 | ||
A similar helical arrangement was observed at temperatures below −40 °C, in the copolymer 145,101 obtained from the two monomers 143 and 144 bearing a C60 unit or a chiral chain, respectively, as side-groups (Scheme 54). The copolymerization was performed using [Rh(nbd)Cl]2 and triethylamine, and a regular cis-transoid polyacetylene chain was formed. The content of fullerene bearing units reflects the composition of the starting mixture of the monomers.
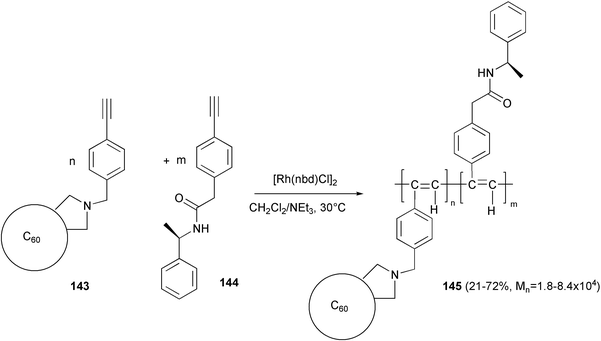 | ||
| Scheme 54 | ||
10. Concluding remarks
This contribution has briefly reviewed the use of organometallic reactions for the synthesis of conjugated oligomers and polymers. The major advantages offered by these methodologies are represented by the possibility of building regio- and stereo-regular conjugated structures with a wide range of functional groups and by the generally mild reaction conditions. Moreover, the organometallic strategy represents the preferential access route to oligomers with a well-defined length of the conjugated system. No single method can be considered of general validity for all types of conjugated systems and a wide complementarity exists between the routes depicted. In any event, the options available appear sufficient to cope with the increasing need for complex structures characterized by electrical and optical properties of special interest in the materials field.11. References
- Handbook of Conducting Polymers, ed. T. A. Skotheim, R. L. Elsenbaumer and J. R. Reynolds, Marcel Dekker Inc., New York, 1998 Search PubMed.
- For an overview on the various applications of conjugated polymers see: P. Chandrasekhar, Conducting Polymers, Fundamentals and Applications. A Practical Approach, Kluwer Academic Publishers, Norwell MA, 1999 Search PubMed.
- T. Ito, H. Shirakawa and S. Ikeda, J. Polym. Sci., Polym. Chem. Ed., 1974, 12, 11 CrossRef CAS.
- G. Natta, G. Mazzanti and P. Corradini, Atti Accad. Naz. Lincei, Cl. Sci. Fis., Mat. Nat. Rend., 1958, 25, 3 Search PubMed.
- H. Shirakawa, F. J. Louis, A. G. MacDiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1977, 578 RSC.
- A. Kraft, A. C. Grimsdale and A. B. Holmes, Angew. Chem., Int. Ed., 1998, 37, 402 CrossRef; U. Mitschke and P. Bäuerle, J. Mater. Chem., 2000, 10, 1471 RSC.
- F. Hide, M. A. Diaz-Garcia, B. J. Schwartz and A. J. Heeger, Acc. Chem Res., 1997, 30, 430 CrossRef CAS; U. Lemmer, A. Haugeneder, C. Kallinger and J. Feldman, in Semiconducting Polymers. Chemistry, Physics and Engineering, ed. G. Hadziioannou and P. F. van Hutten, Wiley-VCH, Weinheim, 2000, p. 309 Search PubMed.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CAS; C. J. Brabec and N. S. Sariciftci, in Semiconducting Polymers. Chemistry, Physics and Engineering, ed. G. Hadziioannou and P. F. van Hutten, Wiley-VCH, Weinheim, 2000, p. 515 Search PubMed.
- Nonlinear Optics of Organic Molecules and Polymers, ed. H. S.Nalwa and S. Miyata, CRC, Boca Raton, 1997 Search PubMed.
- R. A. Wessling, J. Polym. Sci., Polym. Symp., 1985, 72, 55 Search PubMed.
- H. G. Gilch and W. L. Wheelwright, J. Polym. Sci. Part A: Polym. Chem., 1966, 4, 1337 CAS.
- H. Becker, H. Spreitzer, W. Kreuder, E. Kluge, H. Vestweber, H. Schenk and K. Treacher, Synth. Met., 2001, 122, 105 CrossRef CAS.
- For some examples of Wittig polymerization see (a) K. D. Gourley, C. P. Lillya, J. R. Reynolds and J. C. W. Chien, Macromolecules, 1984, 17, 1025 CrossRef CAS; (b) Z.-K. Chen, H. Meng, Y.-H Lai and W. Huang, Macromolecules, 1999, 32, 4351 CrossRef CAS; (c) T. Ahn, S.-Y. Song and H.-K. Shim, Macromolecules, 2000, 33, 6764 CrossRef CAS.
- P. Kovacic and M. B. Jones, Chem. Rev., 1987, 87, 357 CrossRef CAS.
- K. Yoshino, S. Hayashi and R. Sugimoto, Jpn. J. Appl. Phys., 1984, 23, 899 CAS.
- G. Tourillon and F. Garnier, J. Electroanal. Chem., 1982, 135, 173 CrossRef CAS.
- S. Machida, S. Miyata and A. Techagumpuch, Synth. Met., 1989, 31, 311 CrossRef CAS.
- A. G. McDiarmid and A. J. Epstein, in: Science and Applications of Conducting Polymers, ed. W. R. Salaneck, D. T. Clark and E. J. Samuelsen, Adam Hilger, Bristol, 1990, p. 117 Search PubMed.
- S. Bräse and A. de Meijere, in Metal-catalyzed Cross-coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, 1998, p. 99 Search PubMed.
- H. P. Wetzel and K. Müllen, Makromol. Chem., 1990, 191, 2837 CrossRef; H. Martelock, A. Greiner and W. Heitz, Makromol. Chem., 1991, 192, 967 CAS; S. Klingelhöfer, C. Schellenberg, J. Pommerehne, H. Bässler, A. Greiner and W. Heitz, Macromol. Chem. Phys., 1997, 198, 1511 CAS.
- Z. Bao, Y. Cheng, R. Cai and L. Yu, Macromolecules, 1993, 26, 5281 CrossRef CAS.
- Z. Bao, K. R. Amundson and A. J. Lovinger, Macromolecules, 1998, 31, 8647 CrossRef CAS.
- J. A. Mikroyannidis, Macromolecules, 2002, 35, 9289 CrossRef CAS.
- Q. Wang and L. Yu, J. Am. Chem. Soc., 2000, 122, 11806 CrossRef CAS.
- W. You, L. Wang, Q. Wang and L. Yu, Macromolecules, 2002, 35, 4636 CrossRef CAS.
- S.-Y. Song, T. Ahn, H.-K. Shim, I.-S. Song and W.-H. Kim, Polymer, 2001, 42, 4803 CrossRef CAS.
- K. L. Paik, N. S. Baek, H. K. Kim, Y. Lee and K. J. Lee, Thin Solid Films, 2001, 417, 132 CrossRef.
- S. Sengupta and S. Bhattacharyya, J. Chem.Soc., Perkin Trans. 1, 1993, 1943 Search PubMed; K. Kikukawa, K. Nagira, F. Wada and T. Matsuda, Tetrahedron, 1981, 37, 31 CrossRef CAS.
- K. Kikukawa, K. Ikenaga, F. Wada and T. Matsuda, Chem. Lett., 1983, 1337 CAS; K. Ikenaga, K. Kikukawa and T. Matsuda, J. Chem. Soc., Perkin Trans. 1, 1986, 1959 RSC.
- K. Ikenaga, S. Matsumoto, K. Kikukawa and T. Matsuda, Chem. Lett., 1988, 873 CAS.
- S. Sengupta, S. Battacharyya and S. K. Sadhukhan, J. Chem. Soc., Perkin Trans. 1, 1998, 275 RSC; S. Sengupta and S. K. Sadhukhan, J. Chem. Soc., Perkin Trans. 1, 1999, 2235 RSC.
- R. Ancora, F. Babudri, G. M. Farinola, F. Naso and R. Ragni, Eur. J. Org. Chem., 2002, 4127 Search PubMed.
- Electronic Materials: The Oligomer Approach, ed. K. Müllen and G. Wegner, Wiley-VCH, Weinheim, 1998 Search PubMed.
- F. Babudri, G. M. Farinola, F. Naso and D. Panessa, J. Org. Chem., 2000, 65, 1554 CrossRef CAS; F. Babudri, G. M. Farinola, L. C. Lopez, M. G. Martinelli and F. Naso, J. Org. Chem., 2001, 66, 3878 CrossRef CAS.
- G. M. Farinola, V. Fiandanese, L. Mazzone and F. Naso, J. Chem. Soc., Chem. Commun., 1995, 2523 RSC; F. Babudri, G. M. Farinola, V. Fiandanese, L. Mazzone and F. Naso, Tetrahedron, 1998, 54, 1085 CrossRef CAS.
- K. Sonogashira, in Metal-catalyzed Cross-coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, 1998, p. 203 Search PubMed.
- U .H. F. Bunz, Chem. Rev., 2000, 100, 1605 CrossRef CAS.
- J. N. Wilson, S. M. Waybright, K. McAlpine and U. H. F. Bunz, Macromolecules, 2002, 35, 3799 CrossRef CAS.
- R. Fiesel and U. Scherf, Macromol. Rapid. Commun., 1998, 19, 427 CrossRef CAS.
- J. S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 5321 CrossRef CAS.
- R. Deans, J. Kim, M. R. Machacek and T. M. Swager, J. Am. Chem. Soc., 2000, 122, 8565 CrossRef CAS.
- V. Francke, T. Mangel and K. Müllen, Macromolecules, 1998, 31, 2447 CrossRef CAS.
- J. H. Moon and T. M. Swager, Macromolecules, 2002, 35, 6086 CrossRef CAS.
- D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2002, 100, 2537 CrossRef CAS.
- F. Babudri, D. Colangiuli, P. Di Lorenzo, G. M. Farinola, O. Omar Hassan and F. Naso, Chem. Commun., 2003, 130 RSC.
- A. Mori, J. Kawashima, T. Shimada, M. Suguro, K. Hirabayashi and Y. Nishihara, Org. Lett., 2000, 2, 2935 CrossRef CAS.
- B. Erdogan, J. N. Wilson and U. H. F. Bunz, Macromolecules, 2002, 35, 7863 CrossRef CAS.
- A. Suzuki, in Metal-catalyzed Cross-coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, 1998, p. 49 Search PubMed.
- P. Galda and M. Rehahn, Synthesis, 1996, 614 Search PubMed.
- M. B. Goldfinger and T. M. Swager, J. Am. Chem. Soc., 1994, 116, 7895 CrossRef CAS.
- K. Kaeriyama, Y. Tsukahara, S. Negoro, N. Tanigaki and H. Masuda, Synth. Met., 1997, 84, 263 CrossRef CAS.
- A. Izumi, M. Teraguchi, R. Nomura and T. Masuda, Macromolecules, 2000, 33, 5347 CrossRef CAS.
- X. Chen, J.-L. Liao, Y. Liang, M. O. Ahmed, H.-E. Tseng and S.-A. Chen, J. Am. Chem. Soc., 2003, 125, 636 CrossRef CAS.
- J. J. S. Lamba and J. M. Tour, J. Am. Chem. Soc., 1994, 116, 11723 CrossRef CAS.
- F. E. Goodson, T. I. Wallow and B. M. Novak, Macromolecules, 1998, 31, 2047 CrossRef CAS.
- M. Jayakannan, J. L. J. van Dongen and R. A. J. Jansenn, Macromolecules, 2001, 34, 5386 CrossRef CAS.
- T. I. Wallow and B. M. Novak, J. Org. Chem., 1994, 59, 5034 CrossRef CAS.
- G. Sotgiu, M. Zambianchi, G. Barbarella and C. Botta, Tetrahedron Lett., 2002, 58, 2245 CrossRef CAS.
- M. Melucci, G. Barbarella and G. Sotgiu, J. Org. Chem., 2002, 67, 8877 CrossRef CAS.
- F. Koch and W. Heitz, Macromol. Chem. Phys., 1997, 198, 1531 CrossRef CAS.
- T. N. Mitchell, in Metal-catalyzed Cross-coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, 1998, p. 167 Search PubMed.
- Z. Bao, W. K. Chan and L. Yu, J. Am. Chem. Soc., 1995, 117, 12426 CrossRef CAS.
- Q. T. Zhang and J. M. Tour, J. Am. Chem. Soc., 1998, 120, 5355 CrossRef CAS.
- V. Farina, Pure Appl. Chem., 1966, 68, 73.
- H. Meng, D. Tucker, S. Chaffins, Y. Chen, R. Helgeson, B. Dunn and F. Wudl, Adv. Mater., 2003, 15, 146 CrossRef CAS.
- R. Galarini, A. Musco, R. Pontellini, A. Bolognesi, S. Destri, M. Catellani, M. Mascherpa and G. Zhuo, J. Chem. Soc., Chem. Commun., 1991, 364 RSC.
- F. Babudri, S. R. Cicco, G. M. Farinola, F. Naso, A. Bolognesi and W. Porzio, Macromol. Rapid Commun., 1996, 17, 905 CrossRef CAS; A. Bolognesi, C. Botta, F. Babudri, G. M. Farinola, O. Hassan and F. Naso, Synth. Met., 1999, 102, 919 CrossRef CAS; F. Babudri, S. R. Cicco, L. Chiavarone, G. M. Farinola, L. C. Lopez, F. Naso and G. Scamarcio, J. Mater. Chem., 2000, 10, 1573 RSC.
- L. Chiavarone, M. Di Terlizzi, G. Scamarcio, F. Babudri, G. M. Farinola and F. Naso, Appl. Phys. Lett., 1999, 75, 2053 CrossRef CAS.
- F. Babudri, A. Cardone, L. Chiavarone, G. Ciccarella, G. M. Farinola, F. Naso and G. Scamarcio, Chem. Commun., 2001, 1940 RSC.
- T. Cassano, R. Tommasi, F. Babudri, A. Cardone, G. M. Farinola and F. Naso, Opt. Lett., 2002, 27, 2176 CrossRef CAS; F. Babudri, A. Cardone, G. M. Farinola, F. Naso, T. Cassano, L. Chiavarone and R. Tommasi, Macromol. Chem. Phys., 2003, 204, 1621 CrossRef CAS.
- E.-I. Negishi and F. Liu, in Metal-catalyzed Cross-coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, 1998, p. 1 Search PubMed.
- T. Yamamoto, Y. Hayashi and A. Yamamoto, Bull. Chem. Soc. Jpn., 1978, 51, 2091 CAS.
- M. Rehahn, A.-D. Schlüter, G. Wegner and W. J. Feast, Polymer, 1989, 30, 1054 CrossRef CAS.
- M. Kobayashi, J. Chen, T. C. Chung, F. Moraes, A. J. Heeger and F. Wudl, Synth. Met., 1984, 9, 77 CrossRef CAS.
- R. D. McCullough and R. D. Lowe, J. Chem. Soc., Chem. Commun., 1992, 70 RSC.
- T.-A. Chen, X. Wu and R. D. Rieke, J. Am. Chem. Soc., 1995, 117, 233 CrossRef CAS.
- D. R. Greve, J. J. Apperloo and R. A. J. Janssen, Eur. J. Org. Chem., 2001, 3437 CrossRef CAS.
- F. Babudri, D. Colangiuli, G. M. Farinola and F. Naso, Eur. J. Org. Chem., 2002, 2785 CrossRef CAS.
- F. Naso, F. Babudri, D. Colangiuli, G. M. Farinola, T. Quaranta, R. Rella, R. Tafuro and L. Valli, J. Am. Chem. Soc., 2003, 125, 9055 CrossRef CAS.
- T. Yamamoto, A. Morita, Y. Miyazaki, T. Maruyama, H. Wakayama, Z. H. Zhou, Y. Nakamura, T. Kanbara, S. Sasaki and K. Kubota, Macromolecules, 1992, 25, 1214 CrossRef CAS.
- B.-Y. An, Y.-H. Kim, D.-C. Shin, S. Y. Park, H.-S. Yu and S.-K. Kwon, Macromolecules, 2001, 34, 3993 CrossRef CAS.
- C. Ego, D. Marsitzky, S. Becker, J. Zhang, A. C. Grimsdale, K. Müllen, J. D. MacKenzie, C. Silva and R. Friend, J. Am. Chem. Soc., 2003, 125, 437 CrossRef CAS.
- F. Babudri, A. Cardone, G. M. Farinola and F. Naso, Tetrahedron, 1998, 54, 14609 CrossRef CAS.
- F. Babudri, A. R. Cicciomessere, G. M. Farinola, V. Fiandanese, G. Marchese, R. Musio, F. Naso and O. Sciacovelli, J. Org. Chem., 1997, 62, 3291 CrossRef CAS.
- E. Thorn-Csàny, P. Kraxner and A. Strachota, Macromol. Rapid Commun., 1998, 19, 223 CrossRef CAS; E. Thorn-Csàny and P. Kraxner, Macromol. Chem. Phys., 1997, 198, 3827 CrossRef CAS.
- H. Sclick, F. Stelzer, S. Tasch and G. Leising, J. Mol. Catal. A: Chem., 2000, 160, 71 CrossRef.
- J. H. Edwards and W. J. Feast, Polym. Commun, 1980, 21, 595 Search PubMed; J. H. Edwards, W. J. Feast and D. C. Bott, Polymer, 1984, 25, 395 CrossRef CAS.
- V. P. Conticello, D. L. Gin and R. H. Grubbs, J. Am. Chem. Soc., 1992, 114, 9708 CrossRef CAS.
- L. Pu, M. W. Wagaman and R. H. Grubbs, Macromolecules, 1996, 29, 1138 CrossRef CAS; M. W. Wagaman and R. H. Grubbs, Macromolecules, 1997, 30, 3978 CrossRef CAS.
- F. L. Klavetter and R. H. Grubbs, J. Am. Chem. Soc, 1988, 110, 7807 CrossRef CAS; C. B. Gorman, E. J. Ginsburg and R. H. Grubbs, J. Am. Chem. Soc., 1993, 115, 1397 CrossRef CAS.
- K. Weiss, A. Michel, E. Auth, U. H. F. Bunz, T. Mangel and K. Müllen, Angew. Chem., 1997, 36, 506 CAS.
- A. Mortreux and M. Blanchard, J. Chem. Soc., Chem. Commun., 1974, 786 RSC; A. Mortreux, N. Dy and M. Blanchard, J. Mol. Catal., 1976, 1, 101 CrossRef CAS.
- L. Kloppenburg, D. Jones and U. H. F. Bunz, Macromolecules, 1999, 32, 4194 CrossRef CAS; J. N. Wilson, W. Steffen, T. G. McKenzie, G. Lieser, M. Oda, D. Neher and U. H. F. Bunz, J. Am. Chem. Soc., 2002, 124, 6830 CrossRef CAS.
- G. Brizius, N. G. Pschirer, W. Steffen, K. Stitzer, H.-C. zur Loye and U. H. F. Bunz, J. Am. Chem. Soc., 2000, 122, 12453 CrossRef CAS.
- V. Percec, Polym. Bull., 1983, 10, 1 CAS.
- Y. Kishimoto, P. Eckerle, T. Miyatake, M. Kainosho, A. Ono, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1999, 121, 12035 CrossRef CAS.
- B. Ochiai, I. Tomita and T. Endo, Macromol. Rapid Commun., 2001, 22, 1485 CrossRef CAS.
- M. Falcon, E. Farnetti and M. Marsich, J. Organomet. Chem., 2001, 629, 187 CrossRef CAS.
- E. Rivera, M. Belletête, X. X. Zhu, G. Durocher and R. Giasson, Polymer, 2002, 43, 5059 CrossRef CAS.
- A. P. H. J. Schenning, M. Fransen and E. W. Meijer, Macromol. Rapid Commun., 2002, 23, 265 CrossRef CAS.
- T. Nishimura, K. Takatani, S. Sakurai, K. Maeda and E. Yashima, Angew. Chem., Int. Ed., 2002, 41, 3602 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2004 |