Effect of iron on delithiation in LixCo1−yFeyO2. Part 1: in-situ electrochemical and X-ray diffraction study
Michael
Holzapfel†
*ab,
Pierre
Strobel
c,
Céline
Darie
c,
Jonathan
Wright
d,
Mathieu
Morcrette
e,
Eric
Chappel
b and
Michael
Anne
e
aLaboratoire d'Electrochimie et de Physicochimie des Matériaux et des Interfaces, CNRS-INPG-UJF, BP 75, 38402, St. Martin d’Hères, France
bLaboratoire des Champs Magnétiques Intenses, CNRS and MPI für Festkörperforschung, BP 166, 38042, Grenoble cedex 9, France
cLaboratoire de Cristallographie, CNRS, BP 166, 38042, Grenoble cedex 9, France
dEuropean Synchrotron Radiation Facility, BP 220, 38043, Grenoble cedex, France
eLaboratoire de Réactivité et Chimie des Solides, CNRS-UPJV, 80039, Amiens cedex, France
First published on 4th November 2003
Abstract
Structural changes occurring upon electrochemical delithiation in LixCo1−yFeyO2 solid solutions have been examined using in-situ electrochemical X-ray diffraction on plastic batteries, for y
= 0.1, 0.2, 0.4 and 0.7. Two key points are emphasised in this study: (i) the influence of partial iron substitution on the LixCoO2 phase diagram, especially regarding the multiple phase transitions present for 0 ≤
x
≤ 1, (ii) the redox behaviour of iron along with cobalt during the electrochemical process. The y
= 0.1 and y
= 0.2 samples could be totally delithiated and resemble unsubstituted LixCoO2: these systems are two-phase in the range 0.6 < x < 0.9, where two distinct rhombohedral phases coexist, and a specific phase exists in the highly delithiated region. However no ordered analog to Li0.5CoO2 is found. The symmetry of the completely delithiated phases is trigonal (O1, CdI2-type, P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1 space group) and monoclinic (O3, CdCl2-type, C2/m space group) for y
= 0.1 and 0.2, respectively. For LixCo0.6Fe0.4O2, the two-phase regime is maintained down to x
= 0.4, and no new phase is detected down to x
= 0.18. The electrochemical capacities obtained show that iron is involved in the oxidation process. This will be confirmed by EXAFS and XANES measurements, the results of which are published in the second part of this series.
m1 space group) and monoclinic (O3, CdCl2-type, C2/m space group) for y
= 0.1 and 0.2, respectively. For LixCo0.6Fe0.4O2, the two-phase regime is maintained down to x
= 0.4, and no new phase is detected down to x
= 0.18. The electrochemical capacities obtained show that iron is involved in the oxidation process. This will be confirmed by EXAFS and XANES measurements, the results of which are published in the second part of this series.
Introduction
Layered oxide materials LiMO2 are among the most promising materials for application as positive electrode materials in lithium batteries. They crystallise in the rhombohedral α-NaFeO2-structure (space group Rm) which is made up of lithium and transition metal cations occupying the octahedral sites of a cubic close packing of oxide ions in alternate layers. Li is present on the 3a sites, M on the 3b sites, and O on the 6c sites of the trigonal structure. LiCoO2 is the most often used material in practical batteries, as its cycling properties and structural integrity upon cycling are excellent and its preparation is easy,1–3 but the reversible capacity is restricted to about 50% of its theoretical capacity due to irreversible structural transformations and safety precautions regarding the instability of the highly delithiated phase.3 Also the cost and toxicity of cobalt have stimulated research to find adequate cobalt substitutes.
The non-toxicity and low cost of iron could make LiCo1−yFeyO2 an interesting cathode material and indeed, several groups have investigated this system. Synthesis is made by either solid state reaction or hydrothermal reaction,4–9 but in this way only a limited substitution level is accessible. The solid solution range has been recently extended using an indirect route involving ion exchange from NaCo1−yFeyO2 phases.10 Previous structural studies clearly showed that the α-NaFeO2-structure is maintained throughout the whole composition range.4–7,10–16
α-NaFeO2-type LiFeO2 and Fe-rich LiCo1−yFeyO2 are known not to be electrochemically active.11,17,18 The reason for this is not known, as other LiFeO2-modifications that are electrochemically active have been reported.19,20 Two possibilities can be envisaged: the instability of Fe4+ formed upon delithiation, and irreversible structural transformations. The presence of Fe4+ in oxidised LiCo1−yFeyO2 has, however, been indicated by Tabuchi et al.4,16 from XANES and Mössbauer results. The question of Fe4+ is all the more interesting, since tetravalent-metal containing CoO2,21–27 NiO221,25,28,29 and Ni1−yCoyO225,26 layered oxides have been prepared recently by electrochemical deintercalation from the corresponding lithiated oxides LiMO2. These lithium-free dioxides are metastable, as they are very sensitive to reduction, so either extreme care has to be taken at their preparation28,29 or in-situ techniques have to be used.21–25 During lithium deintercalation the layered structure is maintained. As several stable Fe4+-containing oxides are known, especially in the alkaline-earth–Fe–O systems,30,31 it should be possible to stabilise Fe4+ in a Co4+ environment. Moreover, the ionic radius (Ri) compatibility between high-spin Co4+ (Ri = 0.53 Å) and Fe4+ (0.585 Å) is much better than for the trivalent homologues Co3+ (low spin, 0.545 Å) and Fe3+ (high spin, 0.645 Å).32 To address this problem, we undertook in-situ synchrotron X-ray diffraction and absorption experiments during electrochemical delithiation-relithiation of different LiCo1−yFeyO2 samples. The results for the X-ray diffraction are discussed in this paper, whereas the X-ray absorption data will be presented in another paper.33
Experimental
1. Sample preparation
Compounds in the LiCo1−yFeyO2 series were obtained by ion exchange reaction starting from NaCo1−yFeyO2, that were prepared by solid state reaction. For the synthesis of NaCo1−yFeyO2 the starting materials Na2O2 (95%, powder, Fluka), Co3O4 (puriss. p. a., Merck) and Fe2O3 (carbonyl iron oxide, BASF) were thoroughly mixed in an agate mortar in argon atmosphere (glove box MB 150 B–G, O2 and H2O < 1 ppm). A 5% excess of Na2O2 was applied in order to account for volatilisation losses. The powders were heated in corundum boats (Degussit Al 23) in air atmosphere with intermittent grinding and X-ray analysis (XRD, Philips Powder Diffractometer PW1130/00, Cu-Kα-radiation).Different members of the LiCo1−yFeyO2 series were prepared by reacting the NaCo1−yFeyO2 samples two times in an eutectic LiCl/LiNO3 mixture (11.8% LiCl, 88.2% LiNO3, FP: 253 °C) for 6 h. The molar ratio Na compound vs. lithium salt was chosen as 1∶10. The reaction products were washed with methanol (reinst, >99.5%, Merck) and dried in vacuum at room temperature. The LiCo1−yFeyO2 compounds thus prepared were used here to study the corresponding LixCo1−yFeyO2 systems with y = 0.1, 0.2, 0.4 and 0.7. These compounds are abbreviated LCF1, LCF2, LCF4 and LCF7, respectively.
2. In-situ batteries
In-situ X-ray diffraction was carried out in plastic LiCo1−yFeyO2/Li-batteries, which were prepared as follows: typically 70 wt% active material are mixed with 20 wt% PVdF-HFP copolymer binder, 10 wt% SP carbon black and 10% of the total electrode mass weight of dibutylphthalate plasticizer and put into acetone. The resulting slurry is stirred for 30 min and then cast on a glass plate to produce self-standing electrode films. The separator used was a microporous PVdF-membrane (Bellcore). The electrode and separator films were cut into the same size and laminated together (typical electrode surfaces were 2 cm2). The plasticizer was removed by extraction with diethyl ether and the resulting laminates were dried under vacuum and put into a glove-box. The plastic battery was then assembled using the electrode/separator laminate, a lithium counter electrode of the same size and two current collectors: a surface-treated aluminium grid for the cathode side and a copper grid for the anode side. The stacking was assembled, wetted with the electrolyte solution (EC∶DMC = 1∶1, 1 M LiPF6) and sealed into aluminium-metallised plastic bags.343. In-situ X-ray diffraction
The plastic batteries were placed on the BM 16 powder diffraction beamline at ESRF, Grenoble (France). High angular and energy resolution was obtained by placing a curved mirror at grazing incidence before the monochromator in order to vertically collimate the X-ray beam. The diffractometer is equipped with a nine-channel crystal analyser stage which provides high resolution with good statistics.35 The diffraction measurements were carried out at an X-ray energy of 40 keV (λ = 0.3096 Å) in transmission mode. The angular domain was −7 to +33° in 2θ, giving full counting statistics in the range 1 to 25° 2θ, with a counting duration of ca. 16 min per scan.Diffracting in transmission mode through complete flat plastic batteries in the beam yielded Bragg reflections from the positive electrode oxide and the aluminium and copper grids used as current collectors. Fig. 1 shows a typical diffraction pattern for the first pattern recorded for the LCF1 sample. Both Al and Cu give intense and sharp lines, which can be used as internal standards throughout the experiment. The transmission geometry has the advantage to probe the entire thickness of the electrode material,36,37 so that surface effects and the variation of the diffraction angle with the scanning depth are excluded.
 | ||
| Fig. 1 Synchrotron X-ray pattern of LCF1 recorded in-situ at 3.05 V vs. Li/Li+ on a Bellcore-type plastic battery (2–16° 2θ range shown; data were actually collected up to 25° with same statistics). The Rm space group indexation is shown. Al and Cu indicate the aluminium and copper grids reflections. Additional bumps and weak peaks in the 2θ
= 4–5.2° range are due to other components of the assembly, as has been seen previously on this type of plastic battery.42 | ||
A PVC holder with a 2-cm diameter central hole was used to maintain the battery in place during the experiment. The data collected from each of the nine detectors were normalised and summed for each scan. The cell parameters were refined using the pattern matching option of the profile fitting programme Fullprof38 using pseudo-Voigt profile functions.
Results and discussion
1. The LixCoO2 system
Much work has been published about the phase diagram of LixCoO2 upon electrochemical delithiation.21–27 Layered phases are usually described using an abbreviation scheme initially proposed by Delmas,39 where On denotes a structure with octahedral interlayer sites and the presence of n![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) MO2 sheets in the unit cell. In this description, the completely delithiated O1 and O3 types correspond to the CdI2-type (Pm1) and CdCl2-type (Rm) structure, respectively. Starting from LiCoO2
(x
= 1), Reimers and Dahn22 and Ohzuku and Ueda23 found the rapid appearance of a second rhombohedral phase R2
(Rm space group, O3-type) at about x
= 0.9 and the subsequent coexistence of the two phases R1 and R2 until x
≈ 0.75, where R2 disappears. Near x
= 0.50 a new, monoclinic phase with a narrow homogeneity range appears (C2/m space group, O3-type). It corresponds to in-plane 1∶1 ordering of lithium and vacancies, and strictly applies to x
= 0.50 only.22,40 At x < 0.50, it is replaced again by the rhombohedral phase R2.
MO2 sheets in the unit cell. In this description, the completely delithiated O1 and O3 types correspond to the CdI2-type (Pm1) and CdCl2-type (Rm) structure, respectively. Starting from LiCoO2
(x
= 1), Reimers and Dahn22 and Ohzuku and Ueda23 found the rapid appearance of a second rhombohedral phase R2
(Rm space group, O3-type) at about x
= 0.9 and the subsequent coexistence of the two phases R1 and R2 until x
≈ 0.75, where R2 disappears. Near x
= 0.50 a new, monoclinic phase with a narrow homogeneity range appears (C2/m space group, O3-type). It corresponds to in-plane 1∶1 ordering of lithium and vacancies, and strictly applies to x
= 0.50 only.22,40 At x < 0.50, it is replaced again by the rhombohedral phase R2.
The hexagonal a-parameter only shows a slight decrease, ascribed to the slightly smaller size of Co4+ compared to Co3+. On the contrary, c first increases steadily until x = 0.6, then decreases drastically.23 The initial increase in c has been explained by the increasing repulsion between the CoO2 layers due to the weaker screening by the remaining lithium ions. The sharp decrease in c at lower lithium content has been ascribed by Tarascon et al.25 to partial electron transfer from Co4+ to O2−, which is made possible by the similarity between the energies of the Co-3d and O-2p bands. An increase in oxygen O−-character as delithiation proceeds would lower the repulsive forces between the two oxygen layers that are no longer screened from one another, as the van der Waals gap is almost empty (x ≈ 0). One can expect that this effect becomes important for x < 0.5 and above all for very small x-values; the sharp decrease in c indeed begins at x ≈ 0.4.
At the end of charge (x ≈ 0.2) another monoclinic phase appears (C2/m, O1-type). In another study where complete delithiation (x ≈ 0) was reached, a final O1 trigonal phase (CdI2-type structure) was proposed.24 The stability of the O1 structure was ascribed to the weaker electronic repulsion between the oxygen anions in this structure type compared to the O3-type. Upon relithiation, the O1 structure is unstable and immediately transforms into the rhombohedral O3-type, as the presence of lithium ions in the interslab space implies face-sharing of LiO6- and CoO6-octahedra in O1, whereas in the O3 case these octahedra share edges only.
In the study of Tarascon et al.,25 the coexistence between the two rhombohedral phases is extended from x = 0.9 to x = 0.5, and the presence of the intermediate monoclinic phase at x = 0.5 is not observed. The totally delithiated phase was again confirmed to be of O1-type.
Recent results published by Sun and Wang26,27 confirm the structure type of the final delithiated phase, but mention the presence of a third, rhombohedral phase in the two-phase region (x = 0.2–0.5) and discuss again the presence of the monoclinic phase around x = 0.5.
Croguennec et al. have determined that the specific composition Li0.13CoO2 is of an H1–3 type, that is a mixed O1–O3 environment for the lithium ions, present in every second layer.41 The presence of O1-stacking faults leads to some line broadening. For further lithium deintercalation they also find a deintercalated phase of O1-type, but with H1–3-stacking faults.
2. Electrochemical behaviour of LixCo1−yFeyO2
Fig. 2 shows the electrochemical cycling of an LCF1-containing battery in galvanostatic mode. The charge rate, initially set at C/10, was decreased in steps when approaching high potentials, defining regions A–C in Fig. 2 with rates C/15 in B and C/20 in C. D is a 30 min rest time and E the discharge, which was also carried out at C/10 rate. At the end of discharge, the current was interrupted when the potential U reached 3.0 V vs. Li/Li+, and resumed after 30 min if U went back up to more than 3.6 V (region F). | ||
| Fig. 2 Electrochemical cycling behaviour of the LCF1 in-situ battery, shown as a function of time: regions A–F correspond to different current regimes. | ||
As for LixCoO2, the charge curve of LCF1 shows a first plateau at about 3.9 V. An increase in U is then observed (with maximum slope at x ≈ 0.5), followed by a second quasi-plateau at 4.55 V around x = 0.25. A second steeper increase leads to a final stabilisation at 5.1–5.2 V vs. Li/Li+. The only differences with respect to LixCoO2 are (i) slightly higher potentials for comparable x-values, (ii) the absence of any distinct feature at x = 0.5, confirming previous results.11 The discharge occurs about 0.5 V below the charge.
The capacity observed on charge Qc was equivalent to the extraction of 1.034 Li, meaning that a small fraction of Qc was actually consumed in parasitic processes. We noted that the battery bag was indeed slightly inflated at the end of charge, indicating gas evolution due to partial electrolyte oxidation at high voltage. The lithium reintercalation allowed reaching an x value of 0.522.
Other LiCo1−yFeyO2 phases with y = 0.2 (LCF2), 0.4 (LCF4) and 0.7 (LCF7) were charged and discharged in similar conditions. Fig. 3 shows the electrochemical cycling curves for the four compounds investigated. The potential difference ΔUc–d between charge and discharge was found to rise with increasing iron content, indicating a possible increase in electrode inner resistance and/or a lower Li+ diffusion in the host material at higher iron contents. According to Reimers et al.,17 the presence of disordered iron ions may indeed hinder lithium diffusion.
 | ||
| Fig. 3 Comparison of the potential vs. composition curves of the four active materials LCF1, LCF2, LCF4 and LCF7. The asterisk denotes a jump in the current regime. The inhomogeneity in the curves for y = 0.1 and 0.2 at high potentials are also due to the changes in the current regimes. The arrows denote the theoretical limit of oxidation of Co3+ only. | ||
The charge curves show that, even taking into account parasitic oxidation of the electrolyte (which can be assumed to account for x
= 0.05–0.1025,42), the experimental capacities exceed the theoretical values corresponding to the full oxidation of (1 −
y)Co3+. This implies partial oxidation of Fe3+ ions in the electrochemical process. Interestingly, this iron oxidation does not show up as a distinct step in the U(x) curve, unlike the LiMn2−yFeyO4 spinel system, where a separate plateau at 4.8–5.0 V due to the Fe3+/4+ redox step is observed.43–45
3. In-situ X-ray diffraction
Fig. 4 shows the evolution of the X-ray diffraction patterns in the 2θ range 9–13.5° during the in-situ delithiation of LCF1. This angular range includes the Rm oxide phase reflections (015), (107), (018/110) and (113) at about 9.6, 11.4, 12.6 and 13.1° 2θ, respectively. Until about x
= 0.4, a regular shift of the first three lines to lower angles is observed, whereas the (113) line shifts slightly to higher angles. Then the diffraction lines broaden significantly and the pattern shows important changes, with the Rm lines vanishing and being replaced by new diffraction lines. The original pattern reappears upon the subsequent discharge.
 | ||
| Fig. 4 Evolution of a typical part of the synchrotron X-ray pattern of LCF1along a complete charge–discharge cycle. The lithium content values x(Li) are indicated on the right. | ||
A significant line broadening is observed at the beginning of delithiation, as in the case of undoped LixCoO2, where it was related to the coexistence of two rhombohedral (Rm) phases R1 and R2.22,23,25,26 As expected for a two-phase region, the U(x) curve is flat at the beginning of this composition range. However no evidence of a monoclinic distortion of the rhombohedral structure could be found at x
= 0.5. Reimers et al.46 stated that a small amount of 3d-metal doping was sufficient to suppress the Li/vacancy ordering responsible for the formation of this phase, and Tarascon et al.25 explained in this way the absence of the monoclinic distortion at about x
= 0.5 in their study on LixCoO2. We thus ascribe the suppression of the monoclinic distortion to the Fe3+ substitution.
The diffraction pattern of the totally delithiated phase no longer corresponds to the Rm phase, but can be indexed best in the Pm1 space group, i.e. as an O1 phase with cell parameters a
= 2.8181(7)
Å and c
= 4.378(3)
Å. The line shapes and quality of the data for the fully delithiated phase did not allow to perform a full structure refinement. It should be reminded here that this study was not aiming at a redetermination of the structure of the terminal phase, which would have required long acquisition times, but rather at the effect of iron on the LixCo1−yFeyO2 phase diagram as a function of x. A pattern-matching (LeBail's) refinement attempt using a monoclinic model (C2/m)
(Rp: 11.0, Rwp: 16.2, χ2: 5.77) similar to that found by some authors for NiO2,25 gave a poorer fit as the O1 model (Rp: 9.05, Rwp: 12.8, χ2: 3.58). Including a second phase as in Tarascon et al.25
(Rp: 8.49, Rwp: 12.3, χ2: 3.27), gave comparable fits, but the higher number of variables in this case explains the better values for the fit parameters obtained. A single-phase CdI2-type structure thus describes best the most delithiated phase obtained here, Co0.9Fe0.1O2. The H1–3 type for an intermediate lithium-poor composition, as found by Croguennec et al.,41 could not have been identified in the cobalt–iron mixed system.
The evolution of refined lattice parameters as a function of lithium content along a full charge–discharge cycle is shown in Fig. 5. As undoped LixCoO2, LCF1 gives rise to a two-phase region R1 + R2 approximately limited by 0.9 > x > 0.55 upon delithiation, with slightly higher cell parameters, as expected from the larger ionic radius of Fe3+ compared to that of low-spin Co3+ (0.64 vs. 0.53 Å). The a-parameter evolution, however, is clearly affected by the presence of Fe3+, as illustrated by the opposite variation of a(R1) and a(R2) in the two-phase range at x = 0.55–0.9 (see Fig. 5(a)). On the contrary, the c-parameter (Fig. 5(b)) follows the typical bell-shaped variation of LixCoO2, as discussed in section 1. Note the dispersion and high esds for a(R2) values in this range, due to heavy overlap of reflections, resulting in refinement difficulties. Sun et al.26 noted in a recent synchrotron in-situ study of LixCoO2 that the presence of two phases shows up on the (101) reflection, for instance, only as a broadening of an apparently unique peak.
 | ||
| Fig. 5 Evolution of the lattice parameters for LCF1 along a complete charge–discharge cycle as a function of the lithium content x(Li). The c-parameter of the Pm1 phase (O1-type) is multiplied by 3 for easy comparison with O3 phases. Due to large the uncertainties in the determination of the a-lattice parameter of the phase R2, the values were replaced by a straight line at 2.8082 Å. | ||
At x
≈ 0.27, a new O1 phase (CdI2-type, Pm1 space group) with smaller cell parameters appears, forming a new two-phase range with the remaining Rm phase R2. Accordingly, the voltage curve displays a quasi-plateau at x
= 0.30–0.20. In this range, the R2 cell parameters remain constant whereas those of O1 vary steeply: a(O1) increases and c(O1) decrease with decreasing lithium content (see Fig. 5(a) and (b), lower left part). This indicates further lithium extraction from the O1 phase only, while the R2 composition reached its limit and remains constant in this range. The sharp decrease in c(O1) can be explained by a partial electron transfer from Co4+ to O2−, as already explained for LixCoO2.25 This behaviour is not influenced by the presence of the small amount of iron. The latter is significantly larger than that of the undoped CdI2-type homolog (4.24–4.25 Å), again in agreement with the larger size of Fe3+/4+ cations compared to Co3+/4+.
Turning now to relithiation, Fig. 5(a) and (b) show that the structural transformations are reversible at least up to x
= 0.5. However, a systematic shift in x with respect to delithiation is visible in the variation of cell parameters of both phases R2 and O1. This effect can be explained by the “loss” of a fraction of the cell current in electrolyte oxidation or other parasitic reactions at high potentials, as noted earlier.25 The corresponding apparent capacity, estimated best from the shift in a(Pm1)
(Fig. 5(a)), gives Δx
= 0.10 ± 0.02. This value is consistent with those reported in previous in-situ studies using Bellcore-type plastic batteries in this potential range.25,42 The relithiation could not be carried out farther than x
= 0.52 with a potential limit of 3 V, showing that lithium reinsertion is only partially reversible, consistently with previous studies.4,11 Interestingly, both a and c reach approximately their initial values at this point. This behaviour could be due to a partial migration of Fe in the structure in the heavy delithiated range (end of charge), altering the interslab distance and hence the cell parameter evolution on subsequent discharge. However, it is not a redox effect: new EXAFS results, (see Part 2 of this series33), show a concurrent variation of Co–O and Fe–O distances for LCF1 on charge and discharge.
 | ||
| Fig. 6 Evolution of the lattice parameters for LCF2 along a complete charge–discharge cycle as a function of the lithium content x(Li). The monoclinic a- and b-cell parameters have been converted to a pseudo-hexagonal setting as explained in the text. | ||
At lower lithium contents, the behaviour of this sample departs more markedly from that of LCF1. The phase that emerges at the transition around x = 0.25 is best refined with a monoclinic structure model (C2/m space group), and is of O3-type, not of O1-type as found for LixCoO2 and LixCo0.9Fe0.1O2. Its lattice parameters are: a = 4.905(2) Å, b = 2.806(2) Å, c = 13.25(2) Å and β = 91.3(2)°. In addition, the system remains two-phase down to the lowest lithium content reached (x = 0.015), as shown in Fig. 7. As in the LCF1 case, the cell parameters of the R2 phase in this range are constant, indicating a fixed R2 composition and lithium extraction from the O3 phase only. The refinement parameters are: Rp: 8.15, Rwp: 11.2, χ2: 3.26 for the biphase monoclinic solution and Rp: 8.52, Rwp: 11.9, χ2: 3.66 for the biphase O1-type solution).
 | ||
| Fig. 7 Comparison of the synchrotron X-ray diffraction pattern of completely delithiated LCF2 with LCF2 at x
= 0.3 near the Rm
(101) reflection. The double contribution (including a shoulder arising from the Rm phase R2) in delithiated LCF2 at 7.3° 2θ is clearly visible. | ||
The monoclinic a- and b-parameters of the terminal O3 can be converted to the corresponding pseudo-hexagonal lattice using the equation
| ah = (am/√3 + bm)/2 |
As in the LCF1 case, a constant shift in c(R2) between charge and beginning of discharge is observed (see Fig. 6(b), top left part). The capacity related to parasitic oxidation is Δx = 0.12–0.15. This is also the probable cause of the constant cell parameters observed for both phases at the end of charge range (x ≤ 0.15). This is consistent with the plateau in U(x) observed in this region.
 | ||
| Fig. 8 Evolution of the lattice parameters for LCF4 along a complete charge–discharge cycle as a function of the lithium content x(Li). | ||
The major effect of increasing the iron content is in fact to make delithiation thermodynamically more difficult. Fig. 3 shows that the charge curve is shifted by ca. 0.5 V towards higher potentials with respect to LCF1 or LCF2. As a consequence, the charge could not be carried out below x = 0.18, in spite of an excellent electrochemical behaviour of this battery, which could be brought up to 5.4 V without obvious signs of parasitic reactions (no 5.1 V plateau). At this point, the compound should contain at least 0.22 Fe4+. Surprisingly, no specific lithium-poor phase was found in this case, although this appeared for x ≈ 0.25 in both LCF1 and LCF2.
The absence of a delithiated phase of low c-parameter could be due to several factors: (i) the practical impossibility to reach the low x-value necessary for its formation (yet x ≈ 0.25 was sufficient for Fe0.1 and Fe0.2), (ii) a destabilisation of the structure by interslab iron ions or by a too high fraction of Fe4+.33
A partial relithiation was also carried out until x = 0.4. As in the Fe0.1 and Fe0.2 cases, the first rhombohedral phase R1 is not recovered. We also note a steeper variation of both a and c as a function of x(Li) upon relithiation for this sample. This can be explained by the higher fraction of iron ions that show a larger difference in size for their respective 3+ and 4+ oxidation states.
 | ||
| Fig. 9 Evolution of the lattice parameters for LCF7 along a complete charge–discharge cycle as a function of the lithium content x(Li). | ||
 | ||
| Fig. 10 Comparison of the evolution of the lattice parameters for LCF1–LCF4 upon charge. Open and closed symbols correspond to rhombohedral phases R1 and R2, respectively (only the two Rm phases are shown for the three samples). | ||
(i) For all rhombohedral phases but phase R2 in LCF1 (which yielded the least accurate a values), the a-parameter decreases slightly, with an almost constant slope, with decreasing lithium content.
(ii) At the end of delithiation, the a values for the three samples are nearly similar (Fig. 10(a), left side) and very close to those reported for undoped LixCoO2 with x ≈ 0.22,24,25
(iii) The c-parameter evolution shows an important change between 0.2 and 0.4 Fe, for which the typical bell-shape variation vs. lithium known for LixCoO2 disappears.
(iv) At the lithium-poor end of the main two-phase range (x ≈ 0.55 for LCF1 and LCF2, ≈ 0.45 for LCF4), the shift between the cell parameter values for phase R1 and for the continuing phase R2 increases considerably with iron content, and this applies to both a and c parameters.
Observation (ii) shows that the iron content has a large effect on the a parameter of the initial phases containing trivalent Co and Fe (x = 1, Fig. 10(a), right side), but not on the final ones, supporting the presence of cations of similar sizes, hence Co4+ and Fe4+, in the latter (see ionic radii in the introduction). Observation (iv) also supports a much larger effect of iron content on the cell parameters for phase R1 than for phase R2. One can reasonably conclude that the emergence of tetravalent Co and Fe essentially occurs in the R2 phase, whereas the R1 one keeps the memory of a strong Fe3+ effect down to the two-phase range limit.
These results are summarised in a tentative phase diagram as a function of lithium and iron contents in Fig. 11, showing that the general trends of the LixCoO2 system are maintained in LCF1 and LCF2, i.e. a short initial single-phase range followed by a wide two-phase range (R1
+ R2), phase R2 alone, and a new two-phase range at low lithium contents. Note, however, that the ordered compound Li0.5CoO2, detected in several (but not all) studies of the LixCoO2 system, was not found in iron-doped systems, and that a 0.2Fe doping (LCF2) notably alters the phase diagram at low lithium contents: no single-phase at low lithium content is found, in spite of the correct electrochemical behaviour of the battery used, and the final delithiated phase appears to be monoclinic. For 0.4Fe doping (LCF4), the difference is more pronounced, as (i) the first two-phase range extends down to x
≈ 0.45, (ii) no separate, low-lithium content phase was found.
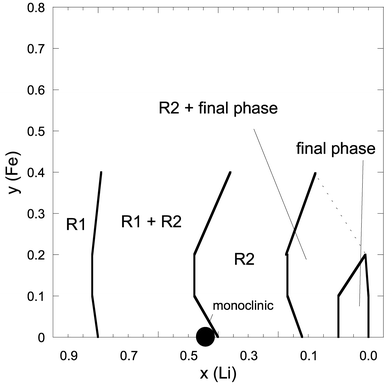 | ||
| Fig. 11 Schematic phase diagram for the LixCo1−yFeyO2-system for 0 ≤ x ≤ 0.4. Range limits for LiCoO2 are approximate, since there are discrepancies in the literature.23–25 | ||
Finally, note that full structural refinements could not be carried out reliably because of unexplained intensity variations. These were also noted on reflections of copper and aluminium, which could be considered as internal standards in these experiments.
Conclusions
The influence of iron substitution on the phase diagram of the LixCo1−yFeyO2 solid solution is addressed in this paper. As for the parent compound LixCoO2 two rhombohedral phases are present in an intermediate composition range (0.9 < x < 0.6), but around x = 0.5 no evidence of a monoclinic distortion could be found. For y = 0.1 and 0.2, at the end of delithiation a structural transformation takes place, with a steep decline in the c lattice parameter. Co0.9Fe0.1O2 shows the same structure as CoO2, i.e. a CdI2-type (O1, Pm1 space group), whereas Co0.8Fe0.2O2 shows a monoclinic distortion and adopts a CdCl2-type structure (O3, C2/m). This is discussed in the light of the Jahn–Teller-effect of Fe4+. LixCo0.6Fe0.4O2 shows a wider two-phase range and does not undergo the structural transition at the end of charge. For all compounds the practical capacity exceeds that calculated for Co3+-oxidation only, which proves the implication of Fe3+-ions in the oxidation process. The structural transformations occurring are partly reversible with reformation of the second rhombohedral phase R2 upon delithiation for the cobalt-rich samples, but the relithiation capacity is relatively poor compared to the delithiation capacity. This study will be completed by an in-situ X-ray absorption investigation of the same samples, which will be described in the second paper of this series.
Acknowledgements
The authors would like to express their gratitude to Dr Yves Chabre for the very helpful discussions and his valuable support. The experiment was carried out under the ESRF proposal number CH 1143.References
- T. Nagaura, 4th International Rechargeable Battery Seminar, Deerfield Beach, FL, USA, 1990 Search PubMed.
- K. Mizushima, P. C. Jones, P. J. Wiseman and J. B. Goodenough, Mater. Res. Bull., 1980, 15, 783 CrossRef CAS.
- T. Ohzuku, A. Ueda, N. Nagayama, Y. Iwakoshi and H. Komori, Electrochim. Acta, 1993, 38, 1159 CrossRef CAS.
- H. Kobayashi, H. Shigemura, M. Tabuchi, H. Sakebe, K. Ado, H. Kageyama, A. Hirano, R. Kanno, M. Wakita, S. Morimoto and S. Nasu, J. Electrochem. Soc., 2000, 147, 960 CrossRef CAS.
- M. Tabuchi, K. Ado, H. Kobayashi, H. Sakaebe, H. Kageyama, C. Masquelier, M. Yonemura, A. Hirano and R. Kanno, J. Mater. Chem., 1998, 9, 199 Search PubMed.
- R. Alcántara, J. C. Jumas, P. Lavela, J. Olivier-Fourcade, C. Pérez-Vicente and J. L. Tirado, J. Power Sources, 1999, 81–82, 547 CrossRef CAS.
- R. Alcantara, P. Lavela, C. Pérez-Vicente, J. L. Tirado, J. Olivier-Fourcade and J. C. Jumas, Solid State Commun., 2000, 115, 1 CrossRef CAS.
- N. Li, J. Li, J. Yang, H. Gao, S. Li and B. Lin, Electrochim. Acta, 2000, 46, 717 CrossRef CAS.
- N. Kalaiselvi, P. Periasami, R. Thirunakaran, B. Ramesh Babu, T. Prem Kumar, N.G. Renganathan, M. Raghavan and N. Muniyandi, Ionics, 2001, 7, 451 Search PubMed.
- M. Holzapfel, C. Haak and A. Ott, J. Solid State Chem., 2001, 156, 470 CrossRef CAS.
- M. Holzapfel, R. Schreiner and A. Ott, Electrochim. Acta, 2001, 46, 1063 CrossRef CAS.
- E. Chappel, M. Holzapfel, G. Chouteau and A. Ott, J. Solid State Chem., 2000, 154, 451 CrossRef CAS.
- E. Chappel, M. Holzapfel, G. Chouteau and A. Ott, J. Magn. Magn. Mater., 2001, 226–230, 652 CrossRef CAS.
- E. Chappel, M. Holzapfel, N. Douakha, G. Chouteau and B. Ouladdiaf, J. Magn. Magn. Mater., 2002, 740, 242.
- N. Douakha, M. Holzapfel, E. Chappel, G. Chouteau, L. Croguennec, A. Ott and B. Ouladdiaf, J. Solid State Chem., 2002, 163, 406 CrossRef CAS.
- H. Sakaebe, H. Shigemura, H. Kobayashi, H. Kageyama and M. Tabuchi, LiBD–Electrode Materials, abstract #71, Arcachon, France, 2001 Search PubMed.
- J. N. Reimers, E. Rossen, C. D. W. Jones and J. R. Dahn, Solid State Ionics, 1993, 61, 335 CrossRef CAS.
- B. Fuchs and S. Kemmler-Sack, Solid State Ionics, 1994, 68, 279 CrossRef CAS.
- R. Kanno, T. Shirane, Y. Inaba and Y. Kawamoto, J. Power Sources, 1997, 68, 145 CrossRef CAS.
- Y. Sakurai, H. Arai, S. Okada and J. Yamaki, J. Power Sources, 1997, 68, 711 CrossRef CAS.
- L. Seguin, G. G. Amatucci, M. Anne, Y. Chabre, P. Strobel, J. M. Tarascon and G. Vaughan, J. Power Sources, 1999, 81–82, 604 CrossRef CAS.
- J. N. Reimers and J. R. Dahn, J. Electrochem. Soc., 1992, 139, 2091.
- T. Ohzuku and A. Ueda, J. Electrochem. Soc., 1994, 141, 2972 CAS.
- G. G. Amatucci, J. M. Tarascon and L. C. Klein, J. Electrochem. Soc., 1996, 143, 1114 CAS.
- J. M. Tarascon, G. Vaughan, Y. Chabre, L. Seguin, M. Anne, P. Strobel and G. G. Amatucci, J. Solid State Chem., 1999, 147, 410 CrossRef CAS.
- X. Sun, X. Q. Yang, J. McBreen, Y. Gao, M. V. Yakovleva, X. K. Xing and M. L. Daroux, J. Power Sources, 2001, 97–98, 274 CrossRef CAS.
- X. Q. Yang, X. Sun and J. McBreen, Electrochem. Commun., 2000, 2, 100 CrossRef CAS.
- L. Croguennec, C. Pouillerie and C. Delmas, Solid State Ionics, 2000, 135, 259 CrossRef CAS.
- L. Croguennec, C. Pouillerie and C. Delmas, J. Electrochem. Soc., 2000, 147, 1314 CrossRef CAS.
- J. B. McChesney, R. C. Sherwood and J. F. Potter, J. Chem. Phys., 1965, 43, 1907 CrossRef CAS.
- S. E. Dann, M. T. Weller and D. B. Currie, J. Solid State Chem., 1991, 92, 237 CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751 CrossRef.
- M. Holzapfel, O. Proux, P. Strobel, C. Darie, M. Borowski and M. Morcrette, J. Mater. Chem., 2004, 14 10.1039/b307672e.
- M. Morcrette, Y. Chabre, G. Vaughan, G. G. Amatucci, J. B. Leriche, S. Patoux, C. Masquelier and J. M. Tarascon, Electrochim. Acta, 2002, 47, 3137 CrossRef CAS.
- J. L. Hodeau, P. Bordet, M. Anne, A. Pratt, A. Fitch, E. Doryhee and G. Vaughan, Powder Diffraction Meeting, Proc. XVII Congress, International Union of Crystallography, Denver, USA, 1996 Search PubMed.
- S. Mukerjee, T. R. Thurston, N. M. Jisrawi, X. Q. Yang, J. McBreen, M. L. Daroux and X. K. Xing, J. Electrochem. Soc., 1998, 145, 466 CAS.
- C. Lampe-Önnerud, T. Gustafsson and J. O. Thomas, Solid State Ionics III, MRS Proceedings, 1992, vol. 293, p. 49 Search PubMed.
- J. Rodriguez-Carvajal, Laboratoire Leon Brillouin (CEA-CNRS), CEA-Saclay, Gif-sur-Yvette, France.
- C. Delmas, C. Fouassier and P. Hagenmuller, Physica B, 1980, 99, 81 CAS.
- Y. Shao-Horn, S. Levasseur, F. Weill and C. Delmas, J. Electrochem. Soc., 2003, 150, A366 CrossRef CAS.
- L. Croguennec, S. Levasseur, M. Morcrette, J. M. Tarascon and C. Delmas, 2002 MRS Fall Meeting, Boston, MA, USA, December 2–6 2002, abstract # EE5.6 Search PubMed.
- M. R. Palacin, F. Le Cras, L. Seguin, M. Anne, Y. Chabre, J. M. Tarascon, G. G. Amatucci, G. Vaughan and P. Strobel, J. Solid State Chem., 1999, 144, 361 CrossRef CAS.
- H. Kawai, M. Nagata, M. Tabuchi, H. Tukamoto and A. R. West, Chem. Mater., 1998, 10, 3266 CrossRef CAS.
- T. Ohzuku, K. Ariyoshi, S. Takeda and Y. Sakai, Electrochim. Acta, 2001, 46, 2327 CrossRef CAS.
- H. Shigemura, H. Sakaebe, H. Kageyama, H. Kobayashi, A. R. West, R. Kanno, S. Morimoto, S. Nasu and M. Tabuchi, J. Electrochem. Soc., 2001, 148, A730 CrossRef CAS.
- J. N. Reimers, J. R. Dahn and U. von Sacken, J. Electrochem. Soc., 1993, 140, 2753.
Footnote |
| † Present address: Paul-Scherrer-Institut, CH-5232 Villigen PSI. E-mail: michael.holzapfel@psi.ch; Fax: +41.(0)56.310.4415; Tel: +41.(0)56.310.2116 |
| This journal is © The Royal Society of Chemistry 2004 |