Synthesis, structure and characterization of two new antimony oxides–LaSb3O9 and LaSb5O12: Formation of LaSb5O12 from the reaction of LaSb3O9 with Sb2O3†
Kang Min
Ok
,
Alex
Gittens
,
Lei
Zhang
and
P. Shiv
Halasyamani
*
Department of Chemistry and Center for Materials Chemistry, 136 Fleming Building, University of Houston, Houston, TX 77204-5003, USA. E-mail: psh@uh.edu
First published on 5th November 2003
Abstract
Two new lanthanum antimony oxides, LaSb3O9 and LaSb5O12, have been synthesized as bulk phase powders and single crystals by solid state reactions using La2O3 (or La(NO3)3·xH2O), Sb2O3 and Sb2O5 as reagents. The structures of LaSb3O9 and LaSb5O12 were determined by single crystal X-ray diffraction. LaSb3O9 contains exclusively Sb5+ cations, whereas LaSb5O12 contains both Sb3+ and Sb5+ cations. LaSb3O9 has a novel three-dimensional framework consisting of edge- and corner-shared Sb5+O6 octahedra. LaSb5O12 exhibits a layered crystal structure consisting of corner-shared Sb5+O6 octahedra. The Sb3+O3 groups cap the SbO6 layers from above and below. The Sb3+ cations are in asymmetric coordination environments attributable to their stereo-active lone-pair. We also report on the reaction of three-dimensional LaSb3O9 with Sb2O3 to form the two-dimensional LaSb5O12. Infrared, thermogravimetric and dielectric analyses are also presented.
Introduction
A rich structural chemistry has been observed in mixed metal oxides containing cations with non-bonded electron pairs (Pb2+, Bi3+, Sb3+, Te4+ and Se4+).1–5 These cations are found in a variety of asymmetric coordination environments attributable to their stereo-active lone-pair. The lone-pair is thought to be the result of a second-order Jahn–Teller (SOJT) distortion.6–10 With these cations, a SOJT distortion reduces the energy between the highest occupied (HOMO) s-orbital and the lowest unoccupied (LUMO) p-orbital through s–p mixing.11–14 Combining this variable coordination geometry with octahedral or tetrahedral moieties produces a great deal of interesting framework architecture. We recently reported the syntheses, structures, and optical behavior of a few materials that contain SOJT distorted cations.15–17 With these ideas in mind, we investigated materials in the La–Sb–oxide system. Attributable to its structural versatility, compounds containing Sb3+ exhibit a great deal of interesting materials properies including magnetic,18 thermoelectric,19 second-harmonic generating,20 and dielectric behavior.21 With respect to rare earth antimony oxides, a few materials have been reported, namely Pr3SbO7,22 La3Li5Sb2O12,23 Ln3Sb5O12 (Ln = Pr, Nd, Sm, Gd, Yb or Eu),24–27 and LaSbO4.28 Except for Ln3Sb5O1224–27 all of these materials contain Sb5+. Structurally the materials are very different, with Pr3SbO722 containing one-dimensional SbO6 chains, whereas La3Li5Sb2O1223 exhibits discrete SbO6 octahedra. Only Ln3Sb5O1224–27 has a three-dimensional structure, consisting of asymmetric SbO3 and SbO4 polyhedra that are linked through oxygen atoms. For LaSbO4, only a powder X-ray diffracation pattern has been reported. In this paper, we report the synthesis, structure and characterization of two new materials, LaSb3O9 and LaSb5O12 (LaSb23+Sb35+O12). In addition, we report on the formation of LaSb5O12 through the reaction of LaSb3O9 and Sb2O3.Experimental
Synthesis
Sb2O3 (Alfa Aesar, 99.6%), Sb2O5 (Aldrich, 99.995%), La(NO3)3·xH2O (Aldrich, 99.9%) and TeO2 (Aldrich, 99%) were used as received. La2O3 (Aldrich, 99.9%) was dried overnight at 1000 °C before being used. α-Sb2O4 was synthesized by heating Sb2O3 in air at 600 °C for 24 h and the purity was confirmed by powder X-ray diffraction.Polycrystalline LaSb3O9 was prepared by standard solid-state techniques. 1.083 g (3.33 × 10−3 mol) of La(NO3)3·xH2O and 1.618 g (5.00 × 10−3 mol) of Sb2O5 were ground thoroughly with an agate mortar and pestle and pressed into a pellet. The pellet was heated to 350 °C for 3 h, and then to 900 °C for 48 h in air and cooled to room temperature with two intermittent re-grindings. The use of La(NO3)3·xH2O was critical in preventing the reduction of Sb5+ to Sb3+. The powder X-ray diffraction pattern on the resultant white powder is in good agreement with the generated pattern from the single crystal data (see ESI†).
Pure LaSb5O12 powder was synthesized by combining 0.362 g (1.11 × 10−3 mol) of La2O3, 0.648 g (2.22 × 10−3 mol) of Sb2O3 and 1.078 g (3.33 × 10−3 mol) of Sb2O5. The mixture was thoroughly ground and pressed into a pellet. The pellet was wrapped in platinum foil, introduced into a fused silica tube that was sealed under vacuum. The tube was heated to 750 °C for 24 h, 850 °C for 48 h, 900 °C for 24 h, and then cooled to room temperature at a rate of 10 °C min−1 with four intermediate re-grindings. The powder X-ray diffraction pattern on the resultant white powder indicated the material was single-phase and in good agreement with the generated pattern from the single crystal data (see ESI†).
Crystallographic determination
A pale yellow block (0.04 × 0.16 × 0.40 mm) for LaSb3O9 and a colorless block (0.12 × 0.15 × 0.20 mm) for LaSb5O12 were used for single crystal data analyses. Room-temperature intensity data were collected on a Siemens SMART diffractometer equipped with a 1K CCD area detector using graphite-monochromated Mo-Kα radiation. A hemisphere of data was collected using a narrow-frame method with scan widths of 0.30° in omega, and an exposure time of 30 s frame−1. The first 50 frames were re-measured at the end of the data collection to monitor instrument and crystal stability. The maximum correction applied to the intensities was <1%. The data were integrated using the Siemens SAINT program,29 with the intensities corrected for Lorentz, polarization, air absorption, and absorption attributable to the variation in the path length through the detector faceplate. Psi-scans were used for the absorption correction on the hemisphere of data. The data were solved and refined using SHELXS-97 and SHELXL-97, respectively.30,31 All of the metal atoms were refined with anisotropic thermal parameters and converged for I > 2σ(I). All calculations were performed using the WinGX-98 crystallographic software package.32 Crystallographic data, atomic coordinates and displacement parameters, and selected bond distances for LaSb3O9 and LaSb5O12 are given in Tables 1–4. The X-ray powder diffraction data were collected on a Scintag XDS2000 diffractometer at room temperature (Cu-Kα radiation, θ–θ mode, flat plate geometry) in the 2θ range 5–60° with a step size of 0.02°, and a step time of 2 s.| Formula | LaSb3O9 | LaSb5O12 |
|---|---|---|
| a R 1 = Σ||Fo| − |Fc||/Σ|Fo|. b w R 2 = [Σw(Fo2 − Fc2)2/Σw(Fo2)2]1/2. | ||
| Formula weight | 648.16 | 939.66 |
| Crystal system | Orthorhombic | Trigonal |
| Space group | Cmcm (no. 63) |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m
(no. 166) m
(no. 166) |
| a/Å | 6.5505(11) | 7.2707(7) |
| b/Å | 8.8941(11) | 7.2707(7) |
| c/Å | 11.8274(17) | 16.4673(12) |
| α/° | 90 | 90 |
| β/° | 90 | 90 |
| γ/° | 90 | 120 |
| V/Å3 | 689.07(17) | 753.89(12) |
| Z | 4 | 3 |
| μ(Mo-Kα)/mm−1 | 17.735 | 17.466 |
| Refl. collected/unique | 2040/450 | 1549/251 |
| R int | 0.0798 | 0.0472 |
| R 1 a [I > 2σ(I)] | 0.0438 | 0.0230 |
| wR 2 b [I > 2σ(I)] | 0.1261 | 0.0596 |
| Atom | x | y | z | U eq a/Å2 | Occupancy |
|---|---|---|---|---|---|
| a U eq is defined as one third of the trace of the orthogonalized Uij tensor. b All oxygen atoms were refined isotropically. | |||||
| La(1) | 0 | 0.2449(1) | 0.75 | 0.012(1) | 1.0 |
| Sb(1) | 0 | 0.5 | 0.5 | 0.007(1) | 1.0 |
| Sb(2) | 0 | 0.1217(1) | 0.4084(1) | 0.007(1) | 1.0 |
| O(1) | −0.1968(10) | 0.6111(6) | 0.4014(5) | 0.010(1)b | 1.0 |
| O(2) | 0 | 0.3336(10) | 0.3849(8) | 0.012(2) | 1.0 |
| O(3) | 0 | 0.1026(10) | 0.5746(8) | 0.008(2) | 1.0 |
| O(4) | 0 | 0.0605(13) | 0.25 | 0.007(2) | 1.0 |
| Atom | x | y | z | U eq a/Å2 | Occupancy |
|---|---|---|---|---|---|
| a U eq is defined as one third of the trace of the orthogonalized Uij tensor. | |||||
| La(1) | 0.3333 | 0.6667 | 0.1667 | 0.007(1) | 1.0 |
| Sb(1) | 0 | 0 | 0.1517(1) | 0.007(1) | 1.0 |
| Sb(2) | 0.1667 | 0.3333 | 0.3333 | 0.006(1) | 1.0 |
| O(1) | 0.2513(10) | 0.1256(5) | 0.3657(3) | 0.010(1) | 1.0 |
| O(2) | 0.1315(5) | 0.2629(10) | 0.2168(3) | 0.011(1) | 1.0 |
| LaSb3O9 | LaSb5O12 | ||
|---|---|---|---|
| Sb(1)–O(1) × 4 | 2.000(6) | Sb(1)–O(2) × 3 | 1.972(6) |
| Sb(1)–O(2) × 2 | 2.011(10) | Sb(2)–O(1) × 4 | 1.968(2) |
| Sb(2)–O(1) × 2 | 1.989(7) | Sb(2)–O(2) × 2 | 1.970(6) |
| Sb(2)–O(2) | 1.906(9) | ||
| Sb(2)–O(3) | 1.972(10) | ||
| Sb(2)–O(3) | 2.005(9) | ||
| Sb(2)–O(4) | 1.951(3) | ||
CCDC reference numbers 218480 and 218481.
See http://www.rsc.org/suppdata/jm/b3/b307496j/ for crystallographic data in CIF or other electronic format.
Characterization
Infrared spectra were recorded on a Matteson FTIR 5000 spectrometer in the 400–4000 cm−1 range, with the sample pressed between two KBr pellets. Thermogravimetric analyses were carried out on a TGA 2950 (TA Instruments). The samples were contained within platinum crucibles and heated at a rate of 10 °C min−1 from room temperature to 900 °C in nitrogen. The dielectric constant (κ) and quality factor (Q = 1/tan![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) δ) measurements were performed using a HP4192A impedance analyzer operating at 1 MHz. Polycrystalline LaSb3O9 and LaSb5O12 were pressed into 1.2 cm diameter and 0.15 cm thick pellets and sintered at 900 °C for 24 h. The pellets had a density 90% of theoretical. A conducting silver paste was applied to the pellet surfaces for electrodes and cured at 400 °C. The temperature dependence of the dielectric constants (TCK) was measured, between −20 and 100 °C, by placing the pellets in a Linkam THMSE600 hot stage.
δ) measurements were performed using a HP4192A impedance analyzer operating at 1 MHz. Polycrystalline LaSb3O9 and LaSb5O12 were pressed into 1.2 cm diameter and 0.15 cm thick pellets and sintered at 900 °C for 24 h. The pellets had a density 90% of theoretical. A conducting silver paste was applied to the pellet surfaces for electrodes and cured at 400 °C. The temperature dependence of the dielectric constants (TCK) was measured, between −20 and 100 °C, by placing the pellets in a Linkam THMSE600 hot stage.
Results and discussion
Structures
LaSb3O9 exhibits a three-dimensional crystal structure consisting of layers of edge- and corner-shared SbO6 octahedra. Each layer is connected through an oxygen atom to form the three-dimensional topology. As seen in Fig. 1(a), six-membered ring channels are formed that run parallel to the a-axis. The La3+ cations reside in these channels. Within each layer, two [Sb(2)O6/2]− octahedra share edges through O(3), whereas an [Sb(1)O6/2]− octahedron shares corners with four [Sb(2)O6/2]− octahedra through O(1) and O(2) (see Fig. 1(b)). The layers are connected by a single oxygen atom, O(4). The Sb–O bond distances lie in the range 1.906(9)–2.008(10) Å. The La3+ cation is in a seven-fold coordination environment with La–O contacts ranging from 2.431(10) to 2.717(12) Å. In connectivity terms, LaSb3O9 can be formulated as consisting of a {3[SbO6/2]−}3− anionic framework with charge balance maintained by the La3+ cation. Bond valence calculations33,34 on LaSb3O9 resulted in values of 2.66 and 5.10 for La3+ and Sb5+, respectively. | ||
| Fig. 1 (a) Ball-and-stick diagram of LaSb3O9 in the bc plane. Note the six-membered ring parallel to the a-axis in which the La3+ cations are found. (b) Ball-and-stick diagram of local environments in LaSb3O9 indicating the connectivity of Sb(1)O6 and Sb(2)O6 units. | ||
LaSb5O12 (LaSb23+Sb35+O12) exhibits a two-dimensional layered crystal structure composed of corner-shared Sb5+O6 octahedra. The asymmetric Sb3+O3 groups cap the Sb5+O6 octahedra from above and below and serve as intra-layer linkers (see Fig. 2). Each Sb5+O6 octahedron is connected to four additional Sb5+O6 octahedra and two Sb3+O3 trigonal pyramids, whereas each Sb3+O3 pyramid is connected to three Sb5+O6 octahedra. The Sb5+–O bond distances lie in the range 1.968(2)–1.970(6) Å, whereas Sb3+–O has a unique bond distance of 1.972(6) Å. The La3+ cation is in a twelve-fold coordination environment with La–O contacts ranging from 2.673(6) to 2.720(6) Å. The non-bonded electron pair on the Sb3+ cation points toward the La3+. In connectivity terms, the structure can be described as a two-dimensional layer of {[Sb3+O3/2]03[Sb5+O6/2]−}3−. Charge neutrality is maintained by the La3+ cation. Bond valence calculations33,34 resulted in values of 2.91, 3.01 and 5.58 for La3+, Sb3+ and Sb5+, respectively.
 | ||
| Fig. 2 Ball-and-stick diagram of LaSb5O12 in the ac plane. Note that the Sb3+O3 groups serve as intra-layer linkers, by capping the Sb5+O6 layer from above and below. | ||
Interestingly, the structural motif of LaSb5O12 is similar to that of A2TeW3O12 (A = Rb or Cs).35 This similarity can be better understood by re-writing LaSb5O12 as LaSb23+Sb35+O12. Thus both LaSb2Sb3O12 and A2TeW3O12 have a “M3O12” sub-structure (M = Sb5+ or W6+), with the three remaining cations in the formula as either “LaSb2” or “A2Te” for LaSb2Sb3O12 and A2TeW3O12, respectively. Both LaSb2Sb3O12 and A2TeW3O12 contain a layer of MO6 octahedra, Sb5+O6 and W6+O6, respectively, that is similar to hexagonal-WO3.36 However, in LaSb2Sb3O12 these layers are anionic, [Sb5+O6/2]−, whereas in A2TeW3O12 the analogous layers are neutral, [W6+O6/2]0. This difference in charge produces different atomic connectivities in the crystal structures. For LaSb2Sb3O12, the Sb3+O3 groups link to the Sb5+O6 layer on both sides, above and below (see Fig. 3), whereas in A2TeW3O12, the Te4+O3 groups connect to the WO6 layer only on one side. This ‘single-sided’ connectivity of the TeO3 groups results in a non-centrosymmetric polar structure, compared with the ‘above and below’ linking of the SbO3 groups that produces a centrosymmetric structure. Between the MO6 layers (M = Sb5+ or W6+) are the La3+ or A+ (A = Rb or Cs) cations, respectively.
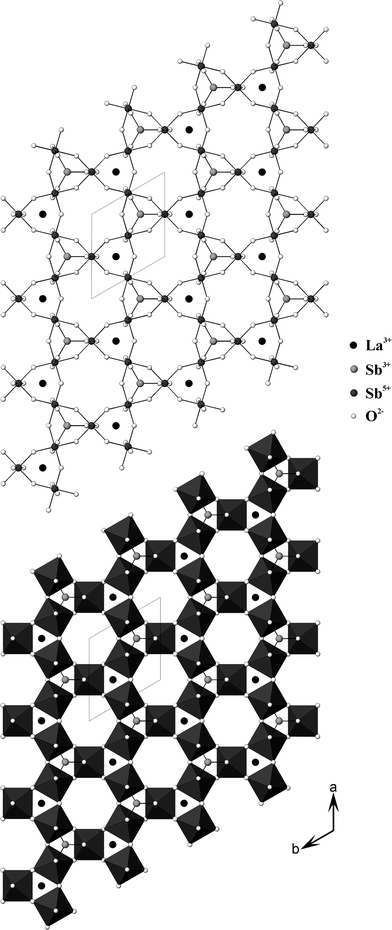 | ||
| Fig. 3 Ball-and-stick (top) and polyhedral (bottom) representation of one-layer of LaSb5O12 in the ab plane. | ||
If we write LaSb5O12 as LaSb2Sb3O12 and ‘subtract’ Sb2O3 from the stoichiometry, we are left with LaSb3O9. Therefore reacting Sb2O3 with LaSb3O9 should produce LaSb5O12. A stoichiometric mixture of LaSb3O9 and Sb2O3 was heated in an evacuated fused silica tube to 900 °C for 24 h and 1000 °C for an additional 24 h. Powder XRD measurements on the reaction product revealed a diffraction pattern consistent with LaSb5O12. Interestingly, this reaction is irreversible. LaSb5O12 is stable up to 1100 °C, with additional heating resulting in decomposition to LaSbO4.28
Characterization
| ν(Sb–O) | ν(Sb–O–Sb) | δ(O–Sb–O) | |
|---|---|---|---|
| LaSb3O9 | 860 | 690 | 484 |
| 804 | 653 | 462 | |
| 736 | 553 | ||
| 526 | |||
| 507 | |||
| LaSb5O12 | 794 | 699 | 489 |
| 755 | 466 | ||
| 422 |
δ) and temperature dependence of the dielectric constant (TCK) for LaSb3O9 are superior to LaSb5O12
(see Table 6). Additional measurements are underway in order to ascertain the origin of this dielectric behavior.
Conclusions
We have synthesized, as both bulk phases and single crystals, two new lanthanum antimony oxides, LaSb3O9 and LaSb5O12. The materials were characterized by single crystal and powder diffraction as well as infrared, thermogravimetric and dielectric analyses. LaSb3O9 has a three-dimensional crystal structure, whereas LaSb5O12 exhibits a layered motif. We have also demonstrated that LaSb5O12 can be formed by the stoichiometric reaction between LaSb3O9 and Sb2O3. We continue to explore the synthesis of new antimony oxides, as well as investigate the incorporation of other cations with stereo-active lone-pairs into new materials.Acknowledgements
We thank the Robert A. Welch Foundation for support. This work was also supported by the NSF-Career Program through DMR-0092054 and an acknowledgment is made to the donors of The Petroleum Research Fund, administered by the American Chemical Society, for partial support of this research. P. S. H. is a Beckman Young Investigator.References
- L. Orgel, J. Chem. Soc., 1959, 3815 RSC.
- G. Meunier and J. Galy, Acta Crystallogr., 1971, 602 CAS.
- J. Galy, G. Meunier, S. Andersson and A. Anstrom, J. Solid State Chem., 1975, 13, 142 CrossRef CAS.
- Y. Arnaud, M. T. Averbuch-Pouchot, A. Durif and J. Guidot, Acta Crystallogr., Sect. B, 1975, 32, 142.
- A. Guesdon and B. Raveau, Chem. Mater., 2000, 12, 2239 CrossRef CAS.
- U. Opik and M. H. L. Pryce, Proc. R. Soc. London A, 1957, 238, 425.
- R. F. W. Bader, Mol. Phys., 1960, 3, 137 CAS.
- R. F. W. Bader, Can. J. Chem., 1962, 40, 1164 CAS.
- R. G. Pearson, J. Mol. Struct.: THEOCHEM, 1983, 103, 25 CrossRef.
- R. A. Wheeler, M.-H. Whangbo, T. Hughbanks, R. Hoffmann, J. K. Burdett and T. A. Albright, J. Am. Chem. Soc., 1986, 108, 2222 CrossRef CAS.
- H. Nikol and A. Vogler, J. Am. Chem. Soc., 1991, 113, 8988 CrossRef CAS.
- H. Nikol and A. Vogler, Inorg. Chem., 1993, 32, 1072 CrossRef CAS.
- G. W. Watson and S. C. Parker, J. Phys. Chem. B., 1999, 103, 1258 CrossRef CAS.
- G. W. Watson, S. C. Parker and G. Kresse, Phys. Rev. B, 1999, 59, 8481 Search PubMed.
- K. M. Ok, N. S. P. Bhuvanesh and P. S. Halasyamani, Inorg. Chem., 2001, 40, 1978 CrossRef CAS.
- Y. Porter, K. M. Ok, N. S. P. Bhuvanesh and P. S. Halasyamani, Chem. Mater., 2001, 13, 1910 CrossRef CAS.
- H.-S. Ra, K. M. Ok and P. S. Halasyamani, J. Am. Chem. Soc., 2003, 125, 7764 CrossRef CAS.
- R. C. T. Slade, G. B. Hix and B. Ducourant, Solid State Ionics, 1997, 99, 233 CrossRef CAS.
- J. S. Kumar, K. V. S. Rao and U. V. S. Rao, J. Less-Common Met., 1990, 161, L19 Search PubMed.
- K. M. Ok, N. S. P. Bhuvanesh and P. S. Halasyamani, J. Solid State Chem., 2001, 161, 57 CrossRef CAS.
- J. S. Kumar, G. Narayana, M. C. Shekar, U. V. S. Rao and V. H. Babu, Phys. Status Solidi A, 1983, 78, 647 CAS.
- J. F. Vent, R. B. Helmholdt and D. J. W. Ijdo, J. Solid State Chem., 1994, 108, 18 CrossRef CAS.
- J. Isasi, M. L. Veiga, A. Jerez and C. Pico, J. Less-Common Met., 1991, 167, 381 Search PubMed.
- A. G. Gukalova, M. N. Tseitlin and K. M. Kurbanov, Zh. Neorg. Khim., 1985, 30, 1978 Search PubMed.
- C. M. Marcano and I. Rasines, Inorg. Chim. Acta, 1985, 109, L15 CrossRef CAS.
- F. Fernandez, R. Saez-Puche, C. Cascales and C. M. Marcano, J. Phys. Chem. Solids, 1989, 50, 871 CrossRef CAS.
- C. Cascales, C. M. Marcano, I. Rasines, F. Fernandez and R. I. Saez-Puche, J. Less-Common. Met., 1989, 149, 63 Search PubMed.
- M. B. Varfolomeev, T. A. Toporenskaya and B. A. Narnov, Zh. Neorg. Khim., 1979, 24, 244 Search PubMed.
- SAINT, Program for Area Detector Absorption Correction, Siemens Analytical X-ray Instruments, Madison, WI, USA, 1995.
- G. M. Sheldrick, SHELXS-97 - A program for automatic solution of crystal structures, Göttingen, Germany, 1997.
- G. M. Sheldrick, SHELXL-97 - A program for crystal structure refinement, Göttingen, Germany, 1997.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B, 1991, 47, 192 CrossRef.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B, 1985, 41, 244 CrossRef.
- J. Goodey, K. M. Ok, J. Broussard, C. Hofmann, F. V. Escobedo and P. S. Halasyamani, J. Solid State Chem., 2003, 175, 3 CrossRef CAS.
- B. Gerand, G. Nowogrocki, J. Guenot and M. J. Figlarz, J. Solid State Chem., 1979, 29, 429 CAS.
- J. A. Alonso, A. Castro, A. Jerez, C. Pico and M. L. Veiga, J. Chem. Soc., Dalton Trans., 1985, 2225 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Powder X-ray diffraction patterns (calculated and experimental). See http://www.rsc.org/suppdata/jm/b3/b307496j/ |
| This journal is © The Royal Society of Chemistry 2004 |