Birnessite-type manganese oxide–alkylamine mesophases obtained by intercalation and their thermal behaviour
Etienne
Wortham
a,
Bernard
Bonnet
a,
Deborah J.
Jones
a,
Jacques
Rozière
*a and
Gary R.
Burns
b
aLaboratoire des Agrégats Moléculaires et Matériaux Inorganiques UMR CNRS 5072, Université Montpellier II, 2 Place Eugène Bataillon, Montpellier cedex 5, 34095, France. E-mail: debtoja@univ-montp2.fr; Fax: +33-4-67 14 33 04; Tel: +33-4-67 14 33 41
bSchool of Chemical and Physical Sciences, Victoria University of Wellington, P. O. Box 600, Wellington, New Zealand
First published on 5th November 2003
Abstract
Layered birnessite-type manganese oxide inserts alkylamines (decylamine–octadecylamine) by ion-exchange and intercalation mechanisms and the concomitant expansion is compatible with formation of a head-to-tail layer of interdigitated amine. The structural arrangement of the MnO2–alkylamine mesophases is sensitive to temperature, and at a temperature dependent on the chain length of the intercalated alkylamine, undergoes a phase transition to a more highly expanded phase. The basal spacing of the more expanded phases formed at higher temperature indicates that de-interdigitation occurs to give a bilayer organisation of neutral and protonated alkylamine species. The phase transition temperature is identified by differential scanning calorimetry. X-Ray absorption spectroscopy shows that while the local structure around manganese is not affected by intercalation, it provides evidence that this transition is accompanied by partial lowering of the oxidation state of manganese and migration of manganese into the interlayer region. This phase transition is reminiscent of some of those observed in phospholipid membranes, but seems to represent a new phenomenon for inorganic intercalates.
Layered ternary manganese oxides AxMnO2 (A = Li, Na, K, …) have attracted recent attention in materials chemistry as battery materials (LiMnO2),1–10 new magnetic materials,11–15 redox catalysts16 and precursors to tunnel-structured manganese oxides.17,18 Birnessite is a common hydrated layered manganese oxide mineral consisting of octahedral edge-shared MnO6 units, with exchangeable cations (Na, K) and water molecules occupying the interlayer region.19–22 Several approaches are known for the preparation of synthetic birnessite, including permanganate oxidation of an Mn(II) salt, reduction of acidifed sodium or potassium permanganate under hydrothermal conditions, or reduction with fumaric acid.23–26 Kinetic and mechanistic features of crystallisation have been reported.27 While the ability of birnessite to incorporate many different metal ions is well known, and their ion-exchange properties have been described,6,21 the formation of inorganic–organic composites has been less well studied. An early report described the formation of an expanded phase by intercalation of dodecylammmonium ion.28 Expanded layered birnessites have since been used as precursor phases in reaction with tetraethoxysilane29 or solutions containing [Al13O4(OH)24·12H2O]7+ to give silica and alumina-pillared manganese oxides,30 respectively. Mesoporous manganese oxide was reported to have been obtained from Mn(OH)2 using hexadecyltrimethylammonium surfactant as a template.31 Delamination has been observed on water-washing ion exchanged tetramethylammonium and tetrabutylammonium–birnessite,32 and tetraalkylammonium colloids have been prepared by reduction of the corresponding permanganate, and the local structure characterised by extended X-ray absorption fine structure spectroscopy.33
In the present work, we carried out a systematic study of the intercalation of potassium birnessite with long chain n-alkylammonium ions CnH2n+1NH3+ (10 ≤ n ≤ 18). Potassium birnessite, and its protonated form, show similar behaviour in this respect to other metal oxides34 or phosphates.35 In addition, however, we have identified novel thermally-induced phase transitions leading to significant further expansion of the basal spacing. The materials have been studied by X-ray diffraction, X-ray absorption spectroscopy, transmission electron microscopy thermal techniques, including thermogravimetric analysis and differential scanning calorimetry.
Experimental
Layered hydrated potassium birnessite (K0.3MnO2) was prepared by hydrothermal decomposition of KMnO4.26 130 ml of a 0.5 M solution of KMnO4 was prepared in a 170 ml sealed Teflon hydrothermal reactor, and its pH was adjusted to 3.5 by adding 1–2 drops of 4 M nitric acid. This solution was subjected to mild hydrothermal treatment at 170 °C for 4 d. The black solid was washed with water several times to eliminate excess KMnO4 until the water ran clear, then recovered by centrifugation. It was dried at room temperature.Intercalation compounds with n-alkylammonium ions (10 ≤ n ≤ 18, n even) were obtained by equilibrating the prepared potassium birnessite with 0.5–1.0 M excess aqueous alkylamine solutions acidified with concentrated hydrochloric acid to pH = 7.36 After contact times ranging from several hours to several days, the solids were recovered, washed several times with a water–ethanol mixture to eliminate the excess amine adsorbed on the surface, and dried in air.
Powder X-ray diffraction patterns were recorded on an automated Philips diffractometer using Cu-Kα radiation (λ = 1.5405 Å). Fe-Kα radiation (λ = 1.9373 Å) was used in variable-temperature X-ray diffraction experiments performed between 20 and 300 °C. Thermogravimetric analysis was carried out in air at a heating rate of 2 °C min−1 using a Stanton-Redcroft STA781 instrument. Elemental analyses for C, H, N, Mn and K were performed by the CNRS Service Central d'Analyse, Vernaison, France. The available oxygen of all samples was determined by the sodium oxalate method, from which the mean oxidation state of manganese was evaluated.37 X-Ray absorption spectra at the manganese K-edge (6539 eV) were recorded in transmission mode at 77 K at the Laboratoire pour l'Utilisation du Rayonnement Electromagnétique, France. X-Ray absorption near edge structure (XANES) spectra were recorded from ca. 50 eV before the edge to 150 eV after the edge in 0.3 eV step-size. In the extended X-ray absorption fine structure (EXAFS) region a step-size of 1.5 eV was used and data recorded to 1000 eV above the absorption edge. He/Ne-filled ionation chambers were used to measure the incident and transmitted X-ray flux. Calibration was carried out prior to all measurements using Mn foil, assigning E0 to the first inflection point in the rising edge. Samples were diluted in boron nitride in proportions adjusted to give an edge jump of ca. 1. After forming a thick mull with paraffin oil, the samples were mounted between the Parafilm windows of stainless-steel sample holders and held at 77 K in a cold-finger cryostat. Experimental data were analysed using the procedures described elsewhere.38,39 Theoretical curves were simulated using functions for the backscattering amplitude and corresponding scattering paths by the tabulated values of McKale and by the ab initio code FEFF-6.40 Multiple scattering effects were computed to be negligible within a radius up to ca. 5 Å from the absorber, and single scattering formalism was used for curve fitting over the first and second atom shells.
Results and discussion
Formation of layered MnO2–alkylammonium mesophases
Elemental and thermogravimetric analysis and the mean oxidation state of manganese in the potassium birnessite prepared lead to the formulation K0.3MnO2·0.65H2O. Its powder X-ray diffraction (XRD) pattern is shown in Fig. 1. It displays three principal diffraction lines corresponding to d-spacings of 7.2, 3.6 and 2.4 Å, characteristic of synthetic alkali-metal containing birnessite with a single sheet of water molecules between MnO2 layers.22 There is a further, less intense peak at 2θ 36.5°, which leads to an estimation of the manganese⋯manganese distance within the layers (ca. 2.85 Å). XRD patterns of the corresponding alkylammonium-intercalates, (CnH2n+1NH3+)xMnO2, are also shown in Fig. 1. These diffraction patterns are all characterised by the presence of multiple higher order reflections, the number of which increases with the number of carbon atoms in the chain (7, 8, 9 and 10 harmonic reflections seen for C10, C12, C14 and C16–C18 intercalates, respectively). The number of higher order reflections observed does not, however, depend upon the degree of crystallinity of the materials, which is, as estimated by the peak width at half-height of the lowest angle diffraction line, similar in each case. | ||
| Fig. 1 XRD patterns of birnessite–alkylamine mesophases: (a) potassium birnessite, (b) decylamine, (c) dodecylamine, (d) tetradecylamine, (e) hexadecylamine, (f) octadecylamine. | ||
For all intercalated phases, another set of diffraction lines is observed at 2θ values between 20 and 24°. The intensity of these diffraction lines is greater for those compounds intercalated with longest-chain alkylamines (see indicated region, Fig. 1(f)), while the position of the lines remains unchanged. The diffraction patterns of alkyamine–birnessite phases are strongly reminiscent of those given by phospholipid membranes41 or liquid crystal inorganic–organic systems such as tin(IV) sulfide–alkylamine composite mesophases.42 It may be concluded that in intercalated birnessite phases also, this group of lines at 2θ 20–24° arises from hydrocarbon chain packing in a liquid crystal-like arrangement in the interlayer region.
The basal spacing of the mesophase alkylammonium intercalates (Table 1) obtained by ion exchange increased linearly from decylammonium (d-spacing 22.5 Å) to octadecylammonium (33.2 Å) by ca. 1.3 Å for each additional –CH2– group in the alkyl chain. This is close to the expected 1.27 Å increase per –CH2– group in an alkyl chain, and indicates that the guest molecules adopt an all-trans configuration. Furthermore, the value of the basal spacing is compatible with a layer of alkylammonium species in which the head groups are organised head-to-tail, leading to an interdigitated arrangement of the hydrocarbon tails, the latter in interaction through hydrophobic van der Waals forces. In summary, the increase in basal spacing for the MnO2–alkylammonium phases is consistent with an monolayer of guest molecules in extended conformation between the manganese oxide layers, and with the carbon chains almost perpendicular to the inorganic layers. The transmission electron micrograph of hexadecylamine-intercalated birnessite, Fig. 2, provides direct observation of a lamellar architecture having an interlayer spacing ca. 30 Å, with the manganese oxide layers appearing non-corrugated and well-aligned. No evidence for other types of arrangement nor of other phases was found.
 | ||
| Fig. 2 Transmission electron micrograph of birnessite–hexadecylamine intercalate: (a) before and (b) after thermal treatment. | ||
| MnO2–alkylamine mesophase | d 001 in low-temperature phase/Å | d 001 in high-temperature phase/Å | Phase transition temperatures from DSC/°C | |
|---|---|---|---|---|
| Hexylamine | — | 22.0 | — | — |
| Decylamine | 22.5 | 32.5 | 25 | 46 |
| Dodecylamine | 25.3 | 37.4 | 46 | 56 |
| Tetradecylamine | 27.8 | 46.0 | 51 | 62 |
| Hexadecylamine | 30.5 | 51.2 | 69 | 70 |
| Octadecylamine | 33.2 | 56.0 | 75 | 86 |
Intercalation leads to materials of composition close to (alkylamine)0.45MnO2, the amount of guest amine exceeding that of potassium initially present (K0.3MnO2). Similar compositions to these were also obtained when aqueous solutions of amines were reacted with proton-form birnessite. Insertion beyond the nominal ion-exchange capacity of the host potassium birnessite suggests that either the insertion reaction is accompanied by partial reduction of manganese, or that non-protonated amine is intercalated along with ion-exchanged alkylammonium species. In tin sulfide–alkylamine composites, Jiang and Ozin42 concluded from 13C and 15N NMR data, that charge balancing protonated amine and neutral amine molecules co-existed in the interlayer region. The amine content surpasses that which can be predicted on the basis of the area requirement of the amine molecule (18.6 Å2) and the available surface area per manganese (7.3 Å2/Mn). For a fully extended conformation of amine molecules perpendicular to the layers, this leads to an expected maximum amine content of 0.39 molecules/Mn. Taken together, the amine content, the increment in the interlayer distance of 1.3 Å/CH2 group, as well as the close-packing on the carbon atoms seen in X-ray diffraction by the characteristic group of lines at 2θ 20–24°, gives a coherent set of observations in favour of an extended arrangement of alkylamine oriented perpendicular to birnessite layers. We may also infer from the stoechiometry that the organisation of the organic component extends beyond that of the individual MnO2 crystallites.
Thermal behaviour of intercalates
On recording X-ray diffraction patterns of the decyl- and dodecyl-amine–birnessite intercalates at various times during the course of the above study at ambient temperature, two different types of pattern were randomly observed. In addition to those described above with basal spacings of 22.5 and 25.3 Å, respectively, a second pattern was obtained, still characteristic of a layered arrangement, but of higher basal spacing. It was realised subsequently that observation of one or other diffraction pattern was related to the temperature at which the material had been stored, those displaying a more expanded structure having experienced a temperature higher than ca. 20 °C. This observation inspired investigation of the influence of temperature on birnessite intercalated with longer chain C14-, C16- and C18-amines. In fact, after heating above 60 °C these systems undergo similar transformation, giving more expanded layered structures having basal spacings collected in Table 1. Apart from the shift to lower angles of all diffraction lines (00l), the patterns of the expanded phases (Fig. 3) are otherwise similar to those of their starting materials, and in each case the line at 2θ 36.5° (related to the Mn⋯Mn distance in the layers) and the grouping of lines at 2θ 20–24°, although broadened, are conserved. These observations allow us to conclude that the manganese oxide sub-lattice remains unchanged, and also that there is still some positional ordering of alkylamines within the interlayer region. | ||
| Fig. 3 XRD patterns of birnessite–alkylamine intercalates after heating at 60 °C for 4 h: (a) decylamine, (b) dodecylamine, (c) tetradecylamine, (d) hexadecylamine, (e) octadecylamine. Lines at 2θ 20–24° correspond to close-packing of alkylamine chains in a liquid crystal-like arrangement. | ||
A plot of basal spacing against the number of carbon atoms shows two sub-sets of points corresponding to C6, C10 and C12 on the one hand and C14, C16 and C18 on the other. The average increment in each case of 2.54 Å per additional –CH2– group and the basal spacings of these more expanded phases are compatible with a double layer of alkylamine in the interlayer region. The origin of the discontinuity between C12 and C14 mesophases is not clear at the moment. To the best of our knowledge, this is the first time that such a transition from an interdigitated monolayer to a bilayer arrangement has been observed for an intercalation compound, although such transitions have been reported for phospholipid membranes and vesicles,43–45 and alkylammonium salts of phosphates,46 tetrahalometallates47etc. Fig. 4 shows the results of variable temperature XRD on the birnessite–hexadecylamine intercalate. The sample was held for 15 min at each chosen temperature before recording the diffraction pattern. It may be seen that lines characteristic of the monolayer start to lose intensity above 40 °C, and that at 60 °C new diffraction lines appear at lower angle corresponding to the presence of the de-interdigitated bilayer phase, stable up to 100 °C. At slightly higher temperature between 100–120 °C, this bilayer phase in turn converts to a third phase, of basal spacing 43 Å. Order in the direction perpendicular to the layers is conserved in this material since a rational series of lines up to sixth order is still seen, however a loss of organisation of the organic sub-lattice is shown by the absence of distinct diffraction lines in the region 2θ 20–24°, Fig. 5. The lower interlayer distance in this third phase indicates inclination of the alkyl chains with respect to the layers. The transmission electron micrograph of Fig. 2(b) shows clear evidence for a periodicity of 42 Å, corresponding to observation of this third phase.
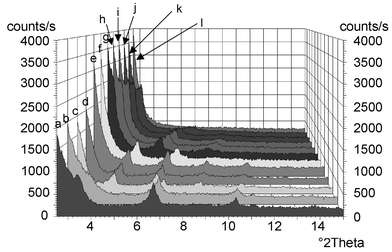 | ||
| Fig. 4 XRD at 20–300 °C of birnessite–hexadecylamine intercalate: (a) 20 °C, (b) 40 °C, (c) 60 °C, (d) 70 °C, (e) 80 °C, (f) 100 °C, (g) 120 °C, (h) 140 °C, (i) 150 °C, (j) 160 °C, (k) 200 °C, (l) 300 °C. | ||
 | ||
| Fig. 5 XRD pattern at room temperature of birnessite–hexadecylamine intercalate after heating to 90 °C. | ||
Differential scanning calorimetry (DSC) traces given by dodecyl-, tetradecyl-, hexadecyl- and octadecyl-amine intercalated birnessite are shown in Fig. 6, and the temperatures of phase transitions are given in Table 1. These experiments were performed on samples held in sealed tubes. As seen in Fig. 6, the trace given during the first heating–cooling cycle differs from those recorded thereafter. For the C12, C14 and C16 mesophases, a weak endothermic peak is observed between 40 and 60 °C in the first heating cycle, followed by two strong exothermic processes between 70 and 100 °C. These two events are not observed on cooling, when two distinct exothermic peaks are seen. The second and third heating–cooling cycles are quite reversible however, with a hysteresis of 10–15 °C between events occurring on heating and on cooling. Such differences were also given by SnS2–mesophase composites,42 where they were attributed to perturbations created by amine-adsorbed CO2. After desorption of CO2 with the first increase in temperature, the transitions are recurring and reversible. For the C12- and C14-intercalates, the first transition in each case involves a large enthalpy change in both directions, endothermic on the heating cycle and exothermic on the cooling cycle. Its narrowness could indicate a first order phase transition and, in agreement with observations in variable-temperature XRD, occurs abruptly. By comparison with the thermal and structural events observed also in lamellar alkylammonium hydrogen phosphates,46 this transition is attributed to the monolayer–bilayer structural modification. A recent study of bulk alkanols with 17 to 20 carbon atoms assigns the first exothermic event in DSC to a solid–solid transition to a rotator phase.48 The more gradual second transition is probably associated with melting of the organic sub-lattice, which then recrystallises again on cooling. The temperature of both transitions increases with the chain length of the inserted amine, and both occur at roughly the same temperature in the MnO2–hexadecylamine intercalate. The sequence of events could be reversed in the MnO2–octadecylamine mesophase. In an open atmosphere, the reverse transformation from bi- to mono-layer phases occurs over several days, which allows X-ray characteration of the bilayer phases even at room temperature.
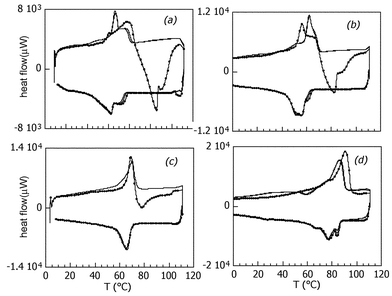 | ||
| Fig. 6 Differential scanning calorimetry curves for birnessite-alkylamine mesophases: (a) dodecylamine, (b) tetradecylamine, (c) hexadecylamine, (d) octadecylamine. First heating–cooling cycle indicated by symbols, second and third heating–cooling cycles: continuous lines. | ||
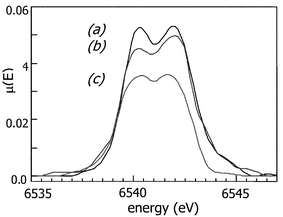
To attempt to elucidate the mechanism of this phase transition, X-ray absorption spectroscopy at the manganese K-edge were performed with parent birnessite,49,50 and intercalated interdigitated and bilayer systems. Normalised near edge spectra of MnO2–hexadeyclamine phases are compared with that of birnessite in Fig. 7. In general, the pre-edge and edge region can provide information on local symmetry and the average oxidation state of the absorbing atom.38,49 The shape of the spectra of Fig. 7 is similar in each case, compatible with an overall retention of the local structure of manganese in the intercalated phases, and the pre-edge feature is characteristic of manganese in an octahedral environment. However, while the position of the rising edge in the XANES of birnessite and the monolayer hexadecylamine intercalate are superimposed, that of the bilayer phase is slightly shifted to lower energy, indicative of a slight lowering of the average manganese oxidation state. Similar results are observed for the tetradecylammonium birnessite and octadecylammonium birnessite phases.
 | ||
| Fig. 7 X-Ray absorption near edge spectra of (a) birnessite, (b) birnessite–hexadecylamine single layer interdigitated intercalate and (c) birnessite–hexadecylamine bilayer intercalate. | ||
Supporting information specific to the valence electronic structure at the manganese centres can be obtained from the pre-edge structures. The pre-edge structures of birnessite, and mono- and bi-layer hexadecylamine phases are compared in Fig. 8, after subtraction of the interpolated tail of the main edge from the spectra of Fig. 7. The pre-edge spectra of birnessite and the interdigitated monolayer phase both have similar intensity, with the two maxima at identical positions, while that of the bilayer is of lower intensity, with the maxima slightly shifted to lower energy. A first explanation for these observations lies in an increase in the MnIII/MnIV ratio, already identified from the shift of the rising edge to lower energy. Other possible factors contributing to a lowering of peak intensity should also be considered however. For example, it has been observed that pre-peak intensity is roughly proportional to the ratio of edge- to corner-sharing MnO6 octahedra, i.e. lower peak intensities and smaller widths are observed for manganese oxides with large tunnel sizes.49 The present evolution in peak intensity and width seems to indicate that the starting birnessite might contain some corner-sharing octahedra in its structure, which are reduced in number in the bilayer intercalated phase.
 | ||
| Fig. 8 Normalised pre-edge region of (a) birnessite, (b) birnessite–hexadecylamine single layer interdigitated intercalate and (c) birnessite–hexadecylamine bilayer intercalate (solid symbols). | ||
The fine structure beyond the absorption edge was extracted using standard methods described elsewhere.38,39Fig. 9 shows that Fourier transformed spectra of birnessite and the monolayer phase spectra are identical both in position and intensity of the maxima, while that of the bilayer phase presents a second shell maximum of lower intensity. This reduction in intensity could be the result either of a lower coordination number, or of a greater distribution of interatomic distances around the average value. The interatomic distances and Debye–Waller factors for the nearest-neighbor Mn–O and Mn–Mn pairs are listed in Table 2. For all three materials, six oxygen atoms lie at an average distance of 1.90 Å from the absorbing manganese atoms. MnO6 octahedra are thus locally affected neither by intercalation nor by the mono- to bi-layer phase transition. In the second shell, the Mn–Mn distance is practically identical in parent birnessite and derived mono- and bi-layer hexadecylamine intercalates but the coordination number is reduced from six in the two former phases, to five in the bilayer material. The similarity between the spectra birnessite and the monolayer intercalate, and their difference with the bilayer intercalate already noted in the XANES region is thus reflected also in the local structure determined by EXAFS, the lower Mn–Mn coordination number being compatible with preferential breakage of Mn–O bonds between corner-shared octahedra. One hypothesis compatible with these results would be the local formation of manganese vacancies in the layers, and a possible migration of manganese atoms to the interlayer space.
 | ||
| Fig. 9 (a) Fourier transformed EXAFS spectra of birnessite (open circles), birnessite–hexadecylamine single layer interdigitated intercalate (solid symbols) and birnessite–hexadecylamine bilayer intercalate (open squares) and (b) example of agreement between experimental (symbols) and calculated (dashed line) curves in k-space for birnessite–hexadecylamine single layer interdigitated intercalate. Spectrum calculated using the numerical data of Table 2. | ||
| N | σ/Å | R/Å | N | σ/Å | R/Å | |
|---|---|---|---|---|---|---|
| a N: coordination number; σ: Debye–Waller factor; R: interatomic distance. Estimated errors are: ±1 on N, ±0.005 Å on σ, 0.02 Å on R. | ||||||
| Birnessite | 6 | 8.0 × 10−2 | 1.90 | 6 | 9.0 × 10−2 | 2.88 |
| Interdigitated monolayer | 6 | 6.2 × 10−2 | 1.90 | 6 | 7.6 × 10−2 | 2.87 |
| De-interdigitated bilayer | 6 | 7.1 × 10−2 | 1.90 | 5 | 8.1 × 10−2 | 2.87 |
Conclusion
Alkylamine-intercalated birnessite de-interdigitates to give materials of significantly expanded interlayer spacing. Although such behavior is observed for phospholipids,41 phosphates,42 or even simple chlorides,51 for inorganic intercalates it is accompanied by a modification of the packing density and composition. Interdigitated structures are favourable when a large space is available for filling by alkyl chains. In smectic lipid bilayers like C10H21NH3Cl, for example, the transition from an interdigitated to a de-interdigitated bilayer has been observed been 40 and 60 °C, and attributed to a partial melting of the alkyl chains.51 The weak interactions such as hydrogen bonds within the “inorganic layers” in such cases can adapt to the modification in the packing of the organic part. In the case of an intercalation compound, the extended character of the inorganic oxide lattice is not favourable to such adjustment. In the present case, the formation of manganese vacancies in the manganese oxide layers, identified by X-ray absorption spectroscopy, could be the triggering mechanism for the de-interdigitation process. In all phases, the periodic arrangement is high, with observation of a rational series of diffraction lines. In addition, for the longer chain alkylamines (C14, C16, C18), a further set of lines suggesting organisation of the organic molecules in the interlayer region is seen. The high organisation has been directly observed by transmission electron microscopy. In common with some other layered structures when intercalated with long-chain alkylamines, contacting the most highly expanded bilayer MnO2 mesophases with liquids such as toluene or trimethylbenzene leads to delamination. The investigation of this property and its use in encapsulation of polymeric species continues in our laboratories.Acknowledgements
We thank the French Ministry of Foreign Affairs, the French Embassy in New Zealand and the Programme International de Coopération Scientifique of the Centre National de la Recherche Scientifique (PICS No. 538) for their long-lasting support of the partnership between Victoria University of Wellington and Université Montpellier II.References
- R. Koksbang, J. Barker, H. Shi and M. Y. Saidi, Solid State Ionics, 1996, 84, 1 CrossRef CAS.
- M. M. Thackeray, Symp. Rechargeable Lithium and Lithium-Ion Batteries 1994 Proceedings, 1995, p. 233 Search PubMed.
- Q. Feng, H. Kanoh, K. Ooi, M. Tani and Y. Nakacho, J. Electrochem. Soc., 1994, 141, L135 CAS.
- C. S. Johnson, D. W. Dees, M. F. Mansuetto, M. M. Thackeray, D. R. Vissers, D. Argyriou, C.-K. Loong and L. Christensen, J. Power Sources, 1997, 68, 570 CrossRef CAS.
- P. Strobel and C. Mouget, Mater. Res. Bull., 1993, 28, 93 CAS.
- P. Le Goff, N. Baffier, S. Bach and J. P. Pereira-Ramos, Mater. Res. Bull., 1996, 31, 63 CrossRef CAS.
- A. R. Armstrong and P. G. Bruce, Nature, 1996, 381, 499 CrossRef CAS.
- A. R. Armstrong, H. Huang, R. A. Jennings and P. G. Bruce, J. Mater. Chem., 1998, 8, 255 RSC.
- S. Bach, J. P. Pereira-Ramos and N. Baffier, Electrochim. Acta, 1993, 38, 1695 CrossRef CAS.
- S. Bach, J. P. Pereira-Ramos and N. Baffier, J. Solid State Chem., 1995, 120, 70 CrossRef CAS.
- Y. Moritomo, A. Asamitsu, H. Kuwahara and Y. Tokura, Nature, 1996, 380, 141 CrossRef CAS.
- Y. Shimakawa, Y. Kubo and T. Manako, Nature, 1998, 392, 473 CrossRef CAS.
- R. Von Helmolt, J. Wecker, B. Holzapfel, L. Schultz and K. Samwer, Phys. Rev. Lett., 1993, 71, 2331 CrossRef CAS.
- M. MacCormack, S. Jin, T. H. Tiefel, R. M. Fleming, J. M. Phillips and R. Ramesh, Appl. Phys. Lett., 1994, 64, 3045 CrossRef CAS.
- C. N. R. Rao, A. K. Cheetham and R. Mahesh, Chem. Mater., 1996, 8, 2421 CrossRef CAS.
- Y. F. Shen, S. L. Suib and C. L. O'Young, J. Catal., 1996, 161, 115 CrossRef CAS.
- Q. Feng, H. Kanoh, Y. Miyai and K. Ooi, Chem. Commun., 1996, 1607 RSC.
- Q. Feng, H. Kanoh and K. Ooi, J. Mater. Chem., 1999, 9, 319 RSC.
- V. A. Drits, E. Silvester, A. I. Gorshkov and A. Manceau, Am. Mineral., 1997, 82, 946 CAS.
- J. E. Post and D. E. Appleman, Am. Mineral., 1990, 75, 477 CAS.
- K. Kuma, A. Usui, W. Paplawsky, B. Gedulin and G. Arrhenius, Miner. Mag., 1994, 58, 425 Search PubMed.
- P. Strobel, J. C. Charenton and M. Lenglet, Rev. Chim. Miner., 1987, 24, 199 Search PubMed.
- S. Bach, M. Henry, N. Baffier and J. Livage, J. Solid State Chem., 1990, 88, 325 CrossRef CAS.
- R. Giovanoli, E. Stähli and W. Feiknecht, Helv. Chim. Acta, 1970, 53, 209 CAS; R. Giovanoli, E. Stähli and W. Feiknecht, Helv. Chim. Acta, 1970, 53, 453 CrossRef CAS.
- J. Luo, A. Huang, S. Park and S. L. Suib, Chem. Mater., 1998, 10, 1561 CrossRef CAS.
- R. Chen, P. Zavalij and M. S. Whittingham, Chem. Mater., 1996, 8, 1275 CrossRef CAS; R. Chen, T. Chirayil, P. Zavalij and M. S. Whittingham, Solid State Ionics, 1996, 86–88, 1 CrossRef CAS.
- J. Luo, Q. Zhang and S. L. Suib, Inorg. Chem., 2000, 39, 741 CrossRef CAS.
- E. Paterson, Am. Mineral., 1981, 66, 424 CAS.
- Z.-H. Liu, K. Ooi, H. Kanoh, W. Tang, X. Yang and T. Tomida, Chem. Mater., 2001, 13, 473 CrossRef CAS.
- S.-T. Wong and S.-F. Cheng, Inorg. Chem., 1992, 31, 1165 CrossRef CAS.
- Z. R. Tian, W. Tong, J. Y. Wang, N. G. Duan, V. V. Krishnan and S. L. Suib, Science, 1997, 276, 926 CrossRef CAS.
- Z.-H. Liu, K. Ooi; W.-P. Tang and T. Tomida, Langmuir, 2000, 16, 4154 CrossRef CAS.
- T. Ressler, S. L. Brock, J. Wong and S. L. Suib, J. Phys. Chem. B, 1999, 103, 6407 CrossRef CAS; S. L. Brock, M. Sanabria, S. L. Suib, V. Urban, P. Thiyagarajan and D. I. Potter, J. Phys. Chem. B, 1999, 103, 7316 CrossRef CAS.
- B. Raveau, Rev. Inorg. Chem., 1987, 9, 37 Search PubMed.
- A. Clearfield and U. Constantino, in Comprehensive Supramolecular Chemistry, ed. G. Alberti and T. Bein, 1996, vol. 7, pp. 107–149 Search PubMed.
- B. Ammundsen, E. Wortham, D. J. Jones and J. Rozière, J. Mol. Cryst. Liq. Cryst., 1998, 311, 327 Search PubMed.
- M. J. Katz, R. C. Clarke and W. F. Nye, Anal. Chem., 1956, 28, 507 CrossRef CAS.
- B. A. Ammundsen, D. J. Jones, J. Rozière and G. R. Burns, Chem. Mater., 1996, 8, 2799 CrossRef CAS.
- P. B. Aitchison, B. A. Ammundsen, D. J. Jones, G. R. Burns and J. Rozière, J. Mater. Chem., 1999, 9, 3125 RSC.
- J. J. Rehr, C. H. Booth, F. Bridges and S. I. Zabinsky, Phys. Rev. B, 1994, 49, 12347 CrossRef CAS.
- I. Hatta, S. Kato and H. Takahashi, Phase Transitions, 1993, 45, 157 Search PubMed.
- T. Jiang and G. A. Ozin, J. Mater. Chem., 1997, 7, 2213 RSC.
- F. S. Hing and G. G. Shipley, Biochemistry, 1995, 34, 11904 CrossRef CAS.
- V. Skita, D. W. Chester, C. J. Oliver, J. G. Turcotte and R. H. Notter, J. Lipid Res., 1995, 36, 1116 Search PubMed.
- S. Maruyama, H. Matsuki, H. Ichimori and S. Kaneshina, Chem. Phys. Lipids, 1996, 82, 125 CrossRef CAS.
- S. R. J. Oliver and G. A. Ozin, J. Mater. Chem., 1998, 8, 1081 RSC.
- F. Neve, A. Crispini, S. Armentano and O. Francescangeli, Chem. Mater., 1998, 10, 1904 CrossRef CAS.
- L. Ventolà, M. Ramírez, T. Calvet, X. Solans, M. A. Cuevas-Diarte, P. Negrier, D. Mondieg, J. C. van Miltenburg and H. A. J. Oonk, Chem. Mater., 2002, 14, 508 CrossRef CAS.
- A. Manceau, A. I. Gorshkov and V. A. Drits, Am. Mineral., 1992, 77, 1133 CAS.
- A. Manceau, A. I. Gorshkov and V. A. Drits, Am. Mineral., 1992, 77, 1144 CAS.
- R. Kind, R. Blinc, H. Arend, P. Muralt, J. Slak, G. Chapuis, K. J. Schenk and B. Zeks, Phys. Rev. A, 1982, 26, 1816 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2004 |