2-Dimensional gel electrophoresis technique for yeast selenium-containing proteins—sample preparation and MS approaches for processing 2-D gel protein spots
Hubert
Chassaigne
*a,
Cyrille C.
Chéry
b,
Guy
Bordin
a,
Frank
Vanhaecke
b and
Adela R.
Rodriguez
a
aEuropean Commission-Joint Research Centre, IRMM, Retieseweg, B-2440 Geel, Belgium. E-mail: hubert.chassaigne@cec.eu.int; Fax: + 32 14 57 17 87
bGhent University, Laboratory of Analytical Chemistry, Proeftuinstraat 86, B-9000 Ghent, Belgium
First published on 28th October 2003
Abstract
A novel integrated approach is proposed for the analysis of intact yeast selenium-containing proteins purified by a gel electrophoresis technique. The strategy consists of three components: high-resolution two-dimensional gel electrophoresis (2-DE) for proteins, laser ablation-inductively coupled plasma-dynamic reaction cell-mass spectrometry (LA-ICP-DRC-MS) for selenium detection and protein characterisation by electrospray mass spectrometry (ion-trap (IT MS) and time-of-flight (TOF MS) instruments). High-resolution 2-DE is the method of choice for protein purification before their characterisation by mass spectrometry techniques. SDS-PAGE which is a denaturating technique is applicable in this case as the selenium is covalently bound to the proteins. LA-ICP-DRC-MS was used, as optimised in previous work for several elements, which also showed that the limit of detection (absolute LOD of 0.5 pg Se per crater or 70 ng Se g−1 gel) was low enough to detect Se in the protein spots. Protein spots were excised from 2-D gels, destained and extracted. Information on the mass of Se-containing proteins were obtained from IT MS and TOF MS. Measurements of the proteins were made with two different MS instruments in order to validate the results and obtain the highest accuracy and resolution on the molecular masses. Up to ten selenium-containing proteins were characterised in terms of molecular mass in the range 9–20 kDa. This strategy has particular application to the possible establishment of a 2-D reference map (Se content and molecular masses) for Se-containing proteins in yeast.
1 Introduction
Food supplements are concentrated sources of nutrients whose purpose is to compensate a low intake of nutrients in the normal diet. They are marketed as pills, tablets, capsules, etc. The European directive 2002/46/EC relating to food supplements establishes harmonised rules for the labelling of such products. The aim is to improve the legislation and to ensure that these products are safe and appropriately labelled so that consumers can make informed choices. The Scientific Committee on Food of the European Commission (SFC) has been asked to review the upper levels of daily intakes of individual minerals that are not likely to have adverse effects on health. However, no information is given concerning the chemical forms of the minerals likely to be present in the food supplements; this might be essential to evaluate the tolerance band of an element.Selenium supplements are often used in order to improve the Se status in the human body. Several producers of Se supplements (containing Se-enriched yeast) are now supplying the European market and there is controversy about the efficiency and the safety of some products. The advantage of yeast is that it metabolises Se in a large variety of organic species and at high doses. Consequently, there is substantial evidence of the complexity of selenium speciation in yeast. A yeast test material has been produced for feasibility studies on selenium species in the context of a share cost action of the European Commission, in particular to enable quantification of inorganic selenium compounds and selenoamino acids and for the investigation of unknown species. The specified concentration range for total selenium in the test material is 1.3 mg g−1. As first step, analytical methods have been developed to allow measurement of inorganic Se, selenomethionine and selenocystine.1 As a second step, a procedure based on polyacrylamide gel electrophoresis (SDS-PAGE) with sensitive Se detection by solid sampling ICP-MS has been proposed for the investigation of the Se-containing proteins.2
The aim of the present research work is to achieve the purification and to evaluate a specific determination method of the Se-containing proteins in order to perform the Se speciation study in yeast material. Two-dimensional gel electrophoresis (2-DE) is being investigated for a complete resolution of Se-containing proteins. 2-DE is the only method currently available which is capable of simultaneously separating hundreds of proteins. The introduction of immobilised pH gradients (IPGs) eliminated the problems of instability and poor sample load capacity associated with carrier ampholyte pH gradients.3 Using the IPGs in a variety of narrow and broad pH range as the first dimension of 2-DE, it is possible to create highly reproducible reference maps, as well as to separate microgram quantities of proteins.4 In the second dimension, a vertical format has a number of advantages over a horizontal format system. Firstly, vertical systems are able to run multiple gels in a single apparatus for reproducibility testing, and secondly, vertical slabs can accommodate high protein loads and allow a higher resolution to be achieved. The objective is the development of a 2-DE method for yeast Se-containing proteins purification.
An essential part of this technology is the possibility of using mass spectrometry for the analysis of gel separated proteins. The method developed by Chéry et al.5 for direct multielemental analysis by laser ablation-inductively coupled plasma-dynamic reaction cell-mass spectrometry (LA-ICP-DRC-MS) is applied to the detection of Se in proteins after 2-DE. Up to now the LA-ICP-MS technique has only been used for the determination of a few elements in 1-D polyacrylamide gels. Two papers have been published on Co in blood serum6 and Pb in humic acid substances.7 The work of Fan et al. on Se determination in soft tissues8 involves the use of a collision cell ICP-MS instrument. The first paper which extends the work from 1-D to 2-D gels and proposes the analysis of other elements is that published by Chéry et al.5 In our case, the ICP-MS is based on the dynamic reaction cell technology (DRC).9
Additionally, this work presents an approach for processing excised protein spots by electrospray mass spectrometry techniques. The critical part is the fastidious sample preparation of excised spots required for further protein analysis by electrospray MS. The most widely used protocol for the confirmation of protein identity following the separation by 1- or 2-DE is the in-gel protein digestion followed by peptide mass fingerprinting by MALDI-TOF MS.10,11 For unknown proteins, peptide sequencing is achieved by LC-MS-MS.12,13 However, in yeast exposed to high doses of selenium it is necessary to use the term selenium-containing proteins and not selenoproteins. Se is incorporated non-specifically into selenoamino acids where it replaces its sulfur analogue. The objective is then not peptide mass fingerprinting or protein sequencing but to achieve the Se speciation in the protein fractions and to determine the accurate mass of each protein. Intact proteins can be extracted for analysis by MS in the case where gel loads exceed ca. 25 pmol.14,15 Individual protein spots are excised from the stained gel by using an automated spot picker system and proteins are extracted in a solvent compatible with electrospray ionisation. The objective is to characterise Se-containing proteins in the 2-D protein map in terms of molecular mass by ion-trap MS (IT MS) and time-of-flight MS (TOF MS). Mass accuracy and resolution obtained for the same proteins with two different techniques are then compared.
2 Experimental
2.1 Chemicals
All chemicals were obtained from Sigma-Aldrich (St. Louis, MO) and were at least analytical reagent grade. Solvents for mass spectrometry were of HPLC grade. Water from a Milli-Q water system (Millipore, Bedford, MA) was used throughout. PlusOne chemicals for 2-D gel electrophoresis were from Amersham Biosciences (Uppsala, Sweden).2.2 Instrumentation
2.3 Analytical procedures
The sample preparation procedure was adapted from previous work.1Fig. 1 shows the complete sample preparation protocol for protein analysis by 2-D gel electrophoresis. The sequential extraction distributes yeast proteins into different fractions based on differential solubility and provides increased resolution of proteins in the resultant 2-D gels. A two step extraction with an aqueous buffer (Tris–HCl buffer) and a SDS-containing buffer (Tris–HCl buffer, 5% SDS) was used. The sample (200 mg of tissue powder) was mixed with 4.0 ml of 10 mM Tris–HCl buffer (pH 8.0) and the suspension was sonicated to disrupt the cells by 10 pulses of 5 s each at maximal energy setting (Step 1 in Fig. 1). Cell disruption was performed at a cold temperature (4 °C) by keeping the sample on ice. The debris were subsequently sedimented by centrifugation at 4380 g for 30 min. The insoluble pellet was then resuspended in 10 mM Tris–HCl buffer containing 5% (w/v) sodium dodecyl sulfate (SDS) (pH 8.0) and submitted to a second extraction step (Step 2 in Fig. 1). The same parameters were used for sonication and centrifugation. The insoluble pellet was then submitted to acidic digestion (HNO3, 65%) for a complete recovery of Se (Step 3). The collected supernatants were kept at 4 °C to protect the sample against proteolysis.
 | ||
| Fig. 1 Protocol for Se extraction, molecular-mass fractionation and sample preparation before the analysis of yeast selenium-containing proteins by 2-dimensional gel electrophoresis (2-DE). | ||
Iodoacetamide (IAM) or iodoacetic acid (Iac) alkylates thiol groups on proteins, preventing their oxidation during 2-D gel electrophoresis.16 To protect selenocysteine (likely to be present in the protein chains) from oxidation, iodoacetic acid was added just prior to use. 1 mol l−1 Iac solution was prepared and its pH was adjusted to 7.0.17Fig. 2 shows the Se carboxymethylation of the amino acid residue Se–Cys with Iac. The derivatisation reaction of the amino acid introduces a mass increase of 58.0055 (Fig. 2). 140 µl Iac solution were added to 2.0 ml yeast extract and incubated at 50 °C for 15 min in the dark. The derivatised extract was submitted to molecular mass cut-off filtration (on 5 kDa cut-off cellulose membranes). The 2 ml extract was poured into the filter cup and the filter unit was centrifuged at 1000 g for 3 h to reduce to a final volume of ca. 200 µl, to free the extract from most of the reagents.
 | ||
| Fig. 2 Se carboxymethylation of the amino acid residue Se–Cys in the protein chain with iodoacetic acid (Iac). Derivatisation creates a mass difference of 58.0055 for the amino acid residue in the protein chain. | ||
Selenium concentration was determined by inductively coupled plasma mass spectrometry (ICP-MS) after sequential extraction and molecular mass cut-off filtration (10 kDa cut-off). Protein concentrations in the extracts were evaluated by using UV spectroscopy at 260 and 280 nm18 to bring the protein concentration compatible with the capacity of 2-D gel electrophoresis.
After isoelectric focusing, the IPG gel strip was prepared for transfer to the second dimension by soaking, with gentle agitation, for 15 min in an equilibration solution (50 mM Tris–HCl pH 8.8, 6 M urea, 30% glycerol, 2% SDS, 0.002% bromophenol blue and DTT). The equilibrated IPG gel strip was embedded at the top of the SDS-PAGE gel in molten 1% (w/v) agarose in cathodic electrode buffer. A set of low molecular mass markers was applied to paper IEF sample application pieces (MW in the range 14–94 kDa). The buffer used for the second dimension SDS-PAGE is a Tris–glycine buffer at high pH, which confers the advantage of minimal protein aggregation and clean separation even at relatively heavy protein loads. Four gels (homogeneous 12.5, 260 × 200 × 1 mm) were run at 120 V per gel for 15 min (30 Vh), then 427 V per gel for 5 h (2135 Vh) with the peltier cooling system at 25 °C.
Two different protocols are used for non-specific detection of protein in 2-D gels: silver and Coomassie blue staining. Silver staining is the most sensitive and allows the detection of less than 1 ng protein per spot. The silver staining protocol gives reproducible results and sensitivity below 1 ng per spot for most proteins. Although 50- to 100-fold less sensitive than silver staining, Coomassie staining is a relatively simple method and preferable when proteins are to be extracted for further characterisation by mass spectrometry (MALDI or electrospray). Among all the Coomassie blue protocols available colloidal staining is recommended, because it shows the highest sensitivity down to 100 ng per protein spot.20
As the actual design of the ablation cell did not allow the presence of the whole gel, pieces of 3.5 cm × 3 cm gel were used for analysis. With laser ablation the formation of craters of 120 µm in protein spots was hardly noticeable and left the majority of the material available for further analysis, especially for the determination of the proteins of interest by ES MS methods. The sensitivity was determined by the ablation of gels hydrated with a known amount of the analyte (Se). The blank used was a polyacrylamide gel soaked in an albumin solution and silver stained, thus containing all components as would a real gel obtained from yeast proteins.5 The standards used were this type of blank gels hydrated in Se standard solutions (resulting concentrations in the range 0.5–10 µg g−1 gel). The limits of detection were calculated with ten ablations at each concentration. LOD for Se was estimated as 0.5 pg per crater (for a 120 µm crater) or as 70 ng Se g−1 gel.5 Although this method is only semi-quantitative with hydrated 2-D gels, the experiments allow the detection of the presence or absence of the selenium in the proteins.
Protocol for intact proteins extracted from 2-D gels. The gels were scanned using Image scanner and analysed using Image Master 2D Elite software (Amersham Biosciences). The position of the selected proteins were exported in a peak list to the Ettan spot picker system. The gels were under liquid in the tray of the spot picker, the camera detects reference markers. Control software converts spot pixel coordinates into picking coordinates (x,y), and the Ettan spot picker selects and transfers gel plugs into 96-well microplates.
Individual Coomassie blue spots were then washed with 15 mM ammomium bicarbonate in MeOH∶H2O, 40∶60, until there was no evidence of remaining Coomassie blue stain. The destaining process completely removes the protein revealer as well as the SDS detergent.14 In the case of silver staining protocol the destaining solution used is a 1∶1 mixture of 15 mM potassium ferricyanide and 50 mM sodium thiosulfate. Acrylamide spots were washed using 100 mM ammonium bicarbonate and Milli-Q water and dehydrated with acetonitrile.
The suspension was sonicated to destroy the gel matrix by 10 pulses of 5 s each at maximal energy setting. Solution composition for extraction is 1% (v/v) formic acid in MeOH∶H2O, 50∶50. The complete protocol is summarised in Fig. 3. The samples were then centrifuged at 16100 g for 5 min before analysis of the supernatant by electrospray MS.
 | ||
| Fig. 3 Sample preparation for MS analysis of intact Se-containing proteins purified by 2-DE technique. The essential stages of the method are summarised. Automated spot picker system is used for gel spot selection and transfer into 96-well microplates. | ||
Electrospray ion-trap MS conditions. The electrospray source was used and protein solutions were injected in the direct infusion mode at 5.0 µl min−1. The instrument was operated in the positive ion mode and a voltage of 3.5 kV was applied on the electrospray needle. The temperature of the heated stainless steel capillary, which was used for desolvation, was 200 °C. Nitrogen was used as a sheath gas and helium as a dampening gas in the ion-trap itself. For MS experiments, proteins were dissolved in solutions of 1% (v/v) formic acid in MeOH∶H2O, 50∶50.
The ion-trap (IT) mass spectrometer was operated in the MS mode for the determination of the molecular mass of molecular ions of proteins. Spectra of proteins were acquired first in the range m/z 500–2000 using a maximum injection time in the trap of 1000 ms during 20 scans (2 min acquisition time). The range was further reduced to m/z 600–1200 to observe the multicharged ions of proteins for accurate mass measurement.
Electrospray time-of-flight MS conditions. The Z-spray ion source was used and protein solutions were injected in the direct infusion mode at 5.0 µl min−1. Proteins were dissolved in a solution of 1% (v/v) formic acid in MeOH∶H2O, 50∶50 for analysis. The instrument was operated in the positive ion mode and a voltage of 3.2 kV was applied at the sprayer. The source and desolvation temperatures were 120 and 250 °C, respectively. The desolvation gas (N2) was set to 350 l h−1.
Time-of-flight (TOF) MS was performed in a continuous extraction mode. In the positive linear mode (V mode), an accelerating voltage of 9.10 kV was used for the TOF. The mass analyser was calibrated with human heart myoglobin (MW range 400–2000). Spectra of proteins were acquired in the range m/z 600–2000 with a scan time of 15 µs and inter scan time of 0.1 s (2.5 min run duration).
3 Results and discussion
3.1 Se quantification after extraction and molecular mass cut-off filtration by ICP-MS
Se concentrations (in µg Se ml−1 solution) were determined after each step of the sample preparation procedure (in the extracts and in the different molecular mass fractions) by quadrupole ICP-MS (Table 1). Se concentrations were determined for 10 independent samples (5 replicates for each measurement). The Se recovery obtained after Tris and SDS extraction (steps 1 and 2 in Fig. 1) were calculated on the basis of the total Se concentration found in the digested yeast material (using HNO3). The value was 1.33 ± 0.17 µg Se g−1 material. Recoveries for Tris and SDS extracts were 19.65 ± 0.55% and 45.27 ± 2.23%, respectively (Table 1). The insoluble pellet was then digested using concentrated HNO3 (65%) for a complete recovery of Se (step 3). A recovery of 91.78 ± 2.10% was obtained for the sequential extraction procedure. 10 kDa cut-off filtration through cellulose membranes provided recoveries of 80.10 ± 2.21% and 83.29 ± 4.62% for Tris and SDS-soluble extracts (cf.Fig. 1), respectively. Low-molecular mass fractions (MW ≤ 10 kDa) were analysed by anion-exchange HPLC-ICP-MS for the quantification of inorganic Se and selenoamino acids (data not shown). Fractions >10 kDa were used for the determination of Se-containing proteins.| Steps | Se concentration in yeast extract/µg ml−1a | Se recovery after sequential extraction (%)a | Se concentration in fraction >10 kDa/µg ml−1b | Se concentration in fraction ≤10 kDa/µg ml−1b | Se recovery after molecular mass fractionation (%)b |
|---|---|---|---|---|---|
| a Results of 10 independent extractions. 5 replicates for each measurement. b Results of 10 independent molecular mass fractionations. 5 replicates for each measurement. | |||||
| 1 Tris extract | 17.54±0.79 | 19.65±1.55 | 39.47±5.44 | 11.99±0.59 | 80.10±2.21 |
| 2 SDS extract | 40.39±2.58 | 45.27±2.23 | 60.00±3.08 | 1.29±0.04 | 83.29±4.62 |
| 3 HNO3 digest | 23.98±1.41 | 26.86±1.82 | — | — | — |
| Total recovery | 91.78±2.10% | ||||
In the Tris extract, the high-molecular weight fraction (MW > 10 kDa) is shown to have a concentration of 39.47 ± 5.44 µg Se ml−1 (fraction reduced to a volume of 200 µl). 10 µl of the Tris extract were used for the sample preparation before the first dimension separation (derivatisation with Iac, 5 kDa filtration and resuspension in IEF buffer as shown in Fig. 1). This corresponds to an amount of selenium of 394 ng on 24 cm IPG gel strips (linear pH range 3–10) or a protein amount of ca. 50 µg after determination by UV spectroscopy. For SDS extract high-molecular weight fraction (MW > 10 kDa) give a concentration of 60.00 ± 3.08 µg Se ml−1 (fraction reduced to 200 µl). In this second case, Se amount loaded on 7 cm IPG gel strip (linear pH range 3–10) is 600 ng and the protein amount was ca. 135 µg. 7 cm gels are used for a rapid screening of the protein content in a fraction whereas the 24 cm gel length is preferred when a higher resolution is required. In the case of the Tris-soluble fraction (water-soluble part of interest in food supplementation) a high efficiency separation has to be achieved before protein spots are selected for LA-ICP-MS and sample preparation for electrospray MS measurements.
3.2 2-D gel electrophoresis and analysis of protein profiles
Appropriate sample preparations are absolutely essential for good 2-D results. Different treatments are required to solubilise different types of proteins in the context of availability studies. The effectiveness of solubilisation depends on the choice of cell disruption method, protein concentration, composition of the sample solution and choice of detergents.23 The protein should be protected from proteolysis during cell disruption, so sample preparation at as low temperature as possible is recommended. Non-protein impurities in the sample (salts, residual buffers, ionic detergents, nucleic acids, polysaccharides, lipids, …) can interfere with separation and subsequent visualisation of the 2-D results, so sample preparation should include steps to rid the sample of these substances. In order to achieve a well-focused first dimension separation, sample proteins must be completely disaggregated and fully solubilised. The sample preparation developed for this work (Fig. 1) provides complete solubilisation of the proteins and results in well focused spots in 2-DE.For yeast Se-containing protein fractions the objectives were the development of a method using 2-D gel electrophoresis to resolve the proteins. Fig. 4 shows a silver-stained 2-D gel with a 7 cm IPG gel strip (linear pH range 3–10) as the first dimension obtained for SDS-soluble proteins (135 µg). It shows that SDS-soluble proteins are in a very wide mass range (10–65 kDa) which confirms the results obtained by 1-D electrophoresis.1 The pI range of the protein spots is between 3 and 8.5. Fig. 5 shows a silver-stained 2-D gel with a 24 cm IPG gel strip (linear pH range 3–10) as the first dimension obtained for Tris-soluble proteins (50 µg). Tris proteins (mainly globular proteins) are in the mass range 10–40 kDa (confirmation of the results obtained for 1-D gels) with a pI range between 3 and 7. In the context of Se yeast supplementation the study is focused on water-soluble Se-containing proteins because of their availability and their nutritional interest.
 | ||
| Fig. 4 2-D map of a SDS-soluble yeast protein extract after silver staining. First dimension run on IPGphor unit using IPG strip pH 3–10, 7 cm. Sample: anodic application of 125 µl of yeast extract containing 135 µg proteins. Running time: 8.5 h, 53.5 kV h. Voltage limitation to 8000 V. Second dimension in 12.5% polyacrylamide gel run on Ettan DALT II system (running time: 5.25 h, 2165 V h). The protein pattern was developed with PlusOne Silver Staining kit protein. | ||
 | ||
| Fig. 5 2-D map of a water-soluble yeast protein extract after silver staining. First dimension run on IPGphor unit using IPG strip pH 3–10, 24 cm. Sample: anodic application of 450 µl of yeast extract containing 50 μg proteins. Running time: 8.5 h, 53.5 kV h. Voltage limitation to 8000 V. Second dimension in 12.5% polyacrylamide gel run on Ettan DALT II system (running time: 5.25 h, 2165 V h). The protein pattern was developed with PlusOne Silver Staining kit protein. | ||
On silver-stained 2-D gel with a wide pH 3–10 linear IPG strip as the first dimension, more than 100 spots can be detected for the protein analysed. However, to improve resolution and sensitivity, narrow-range IPG strips should be used. Such strips have a higher protein loading capacity than standard wide range IPGs24 and consequently, an increased number of protein spots can be observed with the same level of detection sensitivity. A pH range of 3–10 provides an overview of total protein distribution and using a pH range in which some of the proteins have their isoelectric points, i.e. pH 4–7 or 6–9, provides better resolved peaks for these particular proteins. In the case of water-soluble yeast proteins, the objective is to have a specific elemental detection for 2-D electrophoresis to check whether or not, the selenium map matches the whole protein map given by silver-staining. Its is also essential to check the protein purity and to characterise Se-containing proteins in terms of molecular masses before their possible sequencing by MS-MS techniques.
3.3 Detection of selenium in proteins purified by 2-D gel electrophoresis
The limits of detection (LOD) obtained by ETV-ICP-MS for Se determination in protein gels were 50 pg Se per piece of gel (1 mg of gel typically used) or 50 ng Se g−1 gel.2 In order to have a simpler detection method, a laser ablation (LA)-ICP-DRC-MS technique was developed, as described in previous work.5 The use of chemical (dynamic reaction cell technology) resolution in ICP-MS was shown to be essential in order to minimise isobaric interferences for Se isotopes. The volume of a crater in LA is ca. 6 nl whereas the volume of stained spots is ca. 500 nl. LOD for Se were evaluated as 0.5 pg per crater (constant ablation of a cylinder of 1.0 mm height and 120 µm diameter) or 70 ng Se g−1 gel with this technique.5 The detection limits of the two techniques, ETV-ICP-MS and LA-ICP-MS are then comparable. One drawback of LA-ICP-MS is that it takes only a small crater for elemental analysis. We do not have 100% recovery in this case as the main part of the spot is not ablated.5 On the other side, the advantage is that it keeps the majority of the material intact for protein analysis in mass spectrometry. A choice has to be made between quantitation and qualitative detection.Fig. 6 shows an application of the LA-ICP-DRC-MS method to Se determination in 2-D gel of Tris-soluble yeast proteins. 2-D gels were divided in several areas that were introduced independently in the ablation cell. As an example, a part of a gel which was analysed is shown in the insert (3.5 cm × 3 cm) and corresponds to the pI range 4.2–6.2. Spots 1–6 have been ablated and the ICP-DRC-MS signals for 80Se+ are shown in Fig. 6. The amount is evaluated to 1–5 pg Se per crater by external calibration, leading to the conclusion of the presence of Se-containing proteins in the 2-D gel. In spot 6 no signal is observed leading to the conclusion that this protein does not contain Se or that the Se amount is below the LOD of the method. No Se seems to be lost during the separation since no signal is detected at the front and beyond the front of the gel, where low molecular mass oxidation products (such as selenite) would be expected (data not shown).
 | ||
| Fig. 6 Se determination in 2-D gel protein spots by laser ablation-inductively coupled plasma-dynamic reaction cell-mass spectrometry (LA-ICP-DRC-MS). The actual part of the 2-D gel of water-soluble yeast protein extract which was analysed (3.5 cm × 3 cm) is shown in the inset. 80Se+ signal is shown for spots 1–6. | ||
In previous work, identification of Se-containing proteins in yeast was performed using 2-D electrophoresis and radiographic detection after in vivo labelling with 75Se.17 However, it is difficult to compare the applications of the autoradiography technique with that of ETV-ICP-MS (or LA-ICP-MS). These two types of techniques are used for two different applications. In the first case, yeast is grown in the presence of 75Se isotope to get a panoramic view of all selenium-containing proteins. The radiotracer is used to develop the separation method, 2-DE. In our case, the solid sampling-ICP-MS technique is used to measure the natural isotopes of selenium (78Se, 80Se, 82Se) as you can find them in food supplements. The advantages of ETV-ICP-MS are its specificity and sensitivity to measure the natural isotopes of selenium in yeast proteins separated by 1-DE.1 The LA-ICP-DRC-MS technique is much faster and offers good performances such as LOD (70 ng Se g−1 gel), allowing the detection of Se even in the faint spots of 2-DE.5
3.4 Analysis of excised 2-D gels protein spots by electrospray mass spectrometry
Electrospray mass spectrometric methods were considered to go more deeply into the characterisation of Se-containing proteins in yeast. The previously detected protein spots (cf. spot selection in Fig. 6) were used for extraction and analysis in mass spectrometry. As examples to illustrate the high potential of the proposed method, results are described in detail for spots 3–5. Fig. 7a shows the electrospray IT MS spectrum obtained for the extraction of the protein spot 5 (cf. Fig. 6). The mass spectrum was acquired in the range m/z 600–1800 and an envelope of multicharged peaks (+7 to +17) can be seen in the spectrum. The higher the number of m/z signals that can be seen for a given compound in the mass spectrum, the more precise is the result of the molecular mass determination. A deconvoluted mass spectrum is shown in Fig. 7b. Two proteins with very close molecular masses (which differ by a few amino acids in their composition) can seen at 11555.4 ± 0.7 and 11616.3 ± 0.7 Da, respectively. The apparent molecular mass given by the molecular mass markers in 2-DE is less than 14 kDa (Fig. 5). The results are reproducible after confirmation with 5 independent extractions from 2-D gels. Additionally, no degradation product is observed in the MS spectra of proteins leading to the conclusion that fully intact proteins have been extracted.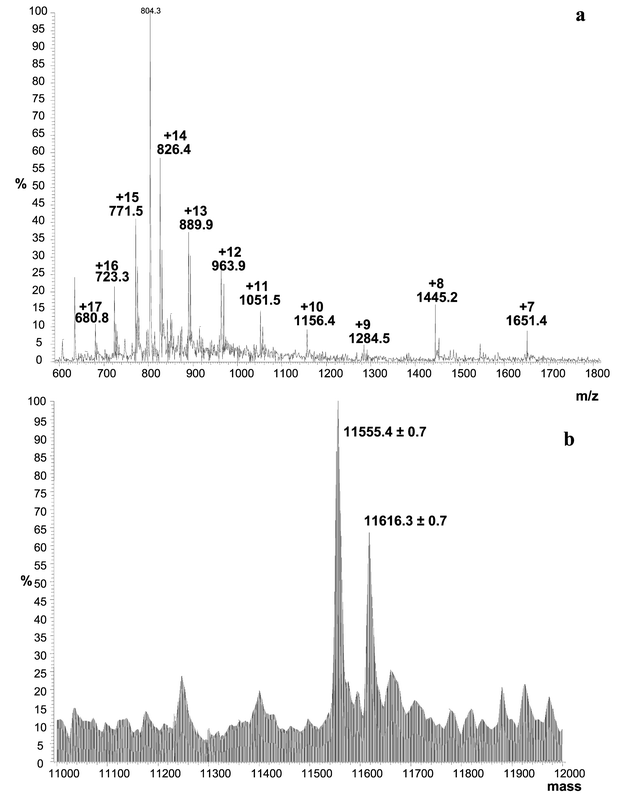 | ||
| Fig. 7 Electrospray IT MS spectrum of intact Se-containing proteins extracted from spot 5 in Fig. 6. Sample preparation described in Fig. 3. (a) MS spectrum in the range m/z 600–1800, (b) deconvoluted mass spectrum in the range 11000–12000 Da. | ||
Fig. 8a shows the electrospray TOF MS spectrum obtained for the extraction of the same spot (number 5 in Fig. 6). An envelope of multicharged peaks (+11 to +16) can be seen in the spectrum and the two neighbouring masses are clearly observed with the high-resolution instrument. Different ionisation profiles obtained with the different techniques (IT and TOF MS) are mainly due to the different ionisation sources used as the same solvent composition is used in both cases. A standard electrospray is used in the case of the ion-trap MS instrument whereas the Q-TOF system is equipped with a Z-spray ionisation system. Two proteins with close masses are determined after deconvolution at 11559.0152 and 11620.1220 (Fig. 8b) as in the case of measurement by IT MS. Higher mass and more accurate values are also obtained with TOF MS than with IT MS. Fig. 9 allows the comparison of the mass accuracy and the resolution obtained by using IT MS and TOF MS instruments. Adjacent peaks in the ion envelope of proteins (from spot 5) obtained in IT MS and TOF MS are shown in Fig. 9a and b, respectively. With the ion-trap system the mass accuracy is ca. 0.01% over the entire mass range (m/z 150–2000).25 The inherent stability of the reflectron TOF analyser routinely delivers excellent mass measurement accuracy (0.0002–0.0005%) for low molecular mass molecules (up to 1000–2000 Da). The typical resolution power of such an instrument is 3000–4000 (FWHM) for masses up to 2000. A TOF instrument also offers 10–100 times more sensitivity than ion-trap instruments. In the case of the analysed proteins (Fig. 9) the gain in sensitivity was evaluated to ca. 45. The high resolving power (5000–10 000 (FWHM)) enables improved mass measurement accuracy for small molecules. In the case of the proteins analysed, the resolutions are much lower and were evaluated to ca. 1000 and 2000 (FWHM) for IT MS and TOF MS instruments, respectively. The significant gains in mass accuracy resolution with the TOF instrument are essential when looking for the sequence of unknown proteins, whereas triple-quadrupole or ion-trap instruments are sufficient for protein confirmation.
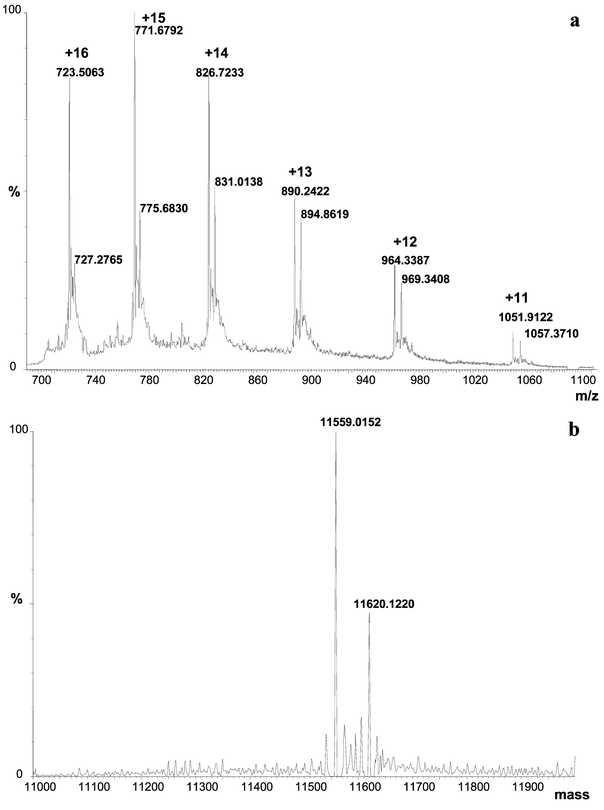 | ||
| Fig. 8 Electrospray TOF MS spectrum of intact Se-containing proteins extracted from spot 5 in Fig. 6. Sample preparation described in Fig. 3. (a) MS spectrum in the range m/z 600–1800, (b) deconvoluted mass spectrum in the range 11000–12000 Da. | ||
 | ||
| Fig. 9 Comparison of MS data obtained for Se-containing proteins with time-of-flight and ion-trap instruments in terms of mass accuracy and resolution. (a) IT MS spectrum of intact Se-containing proteins extracted from spot 5 (Fig. 7). 2 consecutive ionisation sates in the range m/z 810–920, (b) TOF MS spectrum of intact Se-containing proteins extracted from spot 5 (Fig. 8). Two consecutive ionisation states in the range m/z 760–850. | ||
Fig. 10a presents the mass spectrum of spot 3, which is broad and likely to contain more than one protein (cf. Fig. 6). Four Se-containing proteins with very closed molecular masses can be observed at 11559.0150, 11620.0255, 116660.0330 and 11710.0470 Da (Fig. 10b). The two first masses are identical to that found in spot 5 (cf.Fig. 8b) and put emphasis on the possible contamination between spots. The data obtained for the non-resolved spot 4 (data not shown) is similar to the one of spot 3 (Fig. 10) except that the intensity of the four respective peaks is different. We can confirm that the four electrospray signals correspond to four different proteins and not fragments or degradation products as reproducibility is reached over independent experiments. The most performant technique in this case is still electrospray MS. MALDI-TOF MS would not help in the clarification or to have additional information as poor resolution/mass accuracy is obtained for large masses. Up to now 10 intact proteins have been characterised in terms of molecular mass in the range 9–20 kDa (data not shown). Intact proteins with a mass over 20 kDa are difficult to extract quantitatively from the polyacrylamide gel matrix. In this case, it would be necessary to digest the proteins (e.g., trypsin digestion) before extraction and to analyse the peptides by LC-MS-MS.
 | ||
| Fig. 10 Electrospray TOF MS spectrum of intact Se-containing proteins extracted from spot 3 in Fig. 6. Sample preparation described in Fig. 3. (a) MS spectrum in the range m/z 700–1000, (b) deconvoluted mass spectrum in the range 11000–12000 Da. | ||
MALDI-TOF MS is a widely used technique and would, theoretically speaking, allow the atomic selenium isotopic pattern, in the case of selenium-containing peptides resulting from enzymatic digestion of proteins, to be observed. However, because of the contribution of the isotopes of elements other than Se in the molecules, the isotopic pattern of singly charged ions observed differs from that of Se making the interpretation difficult.26 The presence of selenium in intact proteins extracted from the 2-D gel is even more difficult to confirm by MALDI-TOF MS. The resolution and mass accuracy for masses larger than 7–8 kDa do not give a fine structure and only a broad envelope is observed. Consequently no valuable information can be obtained. Intact proteins with masses greater than 20 kDa are also difficult to analyse by MALDI-TOF MS, due to the decreasing signal intensity and mass accuracy. Additionally, a poor transfer of large ions is observed in the case of the Q-TOF instrument used in this work compared to a normal TOF analyser especially dedicated to protein screening in a complex sample and to peptide mass fingerprinting.
4 Conclusion
2-D electrophoresis is essential for establishing high-reproducible reference protein maps for Se-containing proteins. High-resolution 2-D SDS-PAGE is the method of choice for protein purification before their possible characterisation by mass spectrometry (ICP-MS and MALDI electrospray MS). The complete sample preparation and separation methods have been developed in house for the specific application of Se-containing proteins in yeast. This work confirms, as already shown by Chéry et al.,5 that laser ablation-ICP-DRC-MS is a powerful tool in the study of Se-containing proteins, especially when applied to 2-D gels. The advantage of this technique, compared to ETV-ICP-MS, is the lower sample volume and the high throughput. This technique has shown its high potential for the possible establishment of a 2-D reference map (Se content) for Se-containing proteins in yeast material. However, the novelty of this work is mainly due to the complementarity of the techniques used. It combines two MS approaches (ICP-MS and electrospray MS) for processing 2-D gel protein spots and affords valuable information on Se-containing proteins.The accuracy and resolution obtained for the characterisation of the same Se-containing proteins by IT MS and TOF MS have been compared. Ten intact proteins have been measured in the mass range 9–20 kDa. Intact proteins with masses greater than 20 kDa are difficult to extract from the polyacrylamide gels and also to analyse by electrospray MS, due to decreasing signal intensity and mass accuracy for large masses. Future perspectives concern the digestion of Se-containing proteins and peptide extraction followed by their analysis (to determine the amino acid content and evaluate the nutritional value of the proteins) by high-resolution MALDI MS-MS (using the Q-TOF instrument). The hypothesis concerning the presence of selenocysteine in the protein sequence has to be confirmed. Se might be incorporated into selenocysteine and selenomethionine. Additionally, the sequencing is critical since the derivatisation step (with iodoacetic acid) may also affect other amino acids (sulfur-containing amino acids) than the expected selenoamino acids. The difficulty in separating two Se-containing proteins with very close molecular masses, even with a high-resolution 2-D technique, has also to be considered before a complete peptide fragmentation by MS-MS is achieved. In this context, the ensuing work should first include an investigation for the confirmation of the presence of selenocysteine and/or selenomethionine in the protein sequence, before complete sequencing can be achieved.
References
- H. Chassaigne, C. C. Chéry, G. Bordin and A. R. Rodriguez, J. Chromatogr., A, 2002, 976, 409–422 CrossRef CAS.
- C. C. Chéry, H. Chassaigne, L. Verbeeck, R. Cornelis, F. Vanhaecke and L. Moens, J. Anal. At. Spectrom., 2002, 17, 576–580 RSC.
- A. Görg, W. Postel and S. Günther, Electrophoresis, 1988, 9, 531–546 CAS.
- A. Blomberg, L. Blomberg, J. Norbeck, S. J. Fey, P. M. Larsen, M. Larsen, P. Roepstorff, H. Degand, M. Boutry, A. Posch and A. Görg, Electrophoresis, 1995, 16, 1935–1945 CAS.
- C. C. Chéry, D. Günther, R. Cornelis, F. Vanhaecke and L. Moens, Electrophoresis, 2003, 24, 3305–3313 CrossRef.
- J. L. Neilsen, A. Abildtrup, J. Christensen, P. Watson, A. Cox and C. W. McLeod, Spectrochim. Acta, Part B, 1998, 53, 339–345 CrossRef.
- R. D. Evans and J. Villeneuve, J. Anal. At. Spectrom., 2000, 15, 157–161 RSC.
- T. W.-M. Fan, E. Pruszkowski and S. Shuttleworth, J. Anal. At. Spectrom., 2002, 17, 1621–1623 RSC.
- S. D. Tanner, V. I. Baranov and D. R. Bandura, Spectrochim. Acta, Part B, 2002, 57, 1361–1452 CrossRef.
- G. Li, M. Waltham, N. L. Anderson, E. Unsworth, A. Treston and J. N. Weinstein, Electrophoresis, 1997, 18, 391–402 CAS.
- F. Gharahdaghi, C. R. Weinberg, D. A. Meagher, B. S. Imai and S. M. Mische, Electrophoresis, 1999, 20, 601–605 CrossRef CAS.
- L. Marvin, A. Millar, V. Saulot, N. Machour, R. Charlionet, F. Tron and C. Lange, Rapid Commun. Mass Spectrom., 2000, 14, 1287–1292 CrossRef CAS.
- E. P. Romijn, J. Krijgsveld and A. J. R. Heck, J. Chromatogr., A, 2003, 1000, 589–608 CrossRef CAS.
- S. L. Cohen and B. T. Chait, Anal. Biochem., 1997, 247, 257–267 CrossRef CAS.
- H. Ehring, S. Stromberg, A. Tjernberg and B. Noren, Rapid Commun. Mass Spectrom., 1997, 11, 1867–1873 CrossRef CAS.
- A. Görg, W. Postel, J. Weser, S. Günther, J. R. Strahler, S. M. Hanash and L. Somerlot, Electrophoresis, 1987, 8, 122–124 CAS.
- C. C. Chéry, E. Dumont, R. Cornelis and L. Moens, Fresenius'J. Anal. Chem., 2001, 371, 775–781 CrossRef CAS.
- E. Layne, Methods Enzymol., 1957, 3, 447–455 CrossRef.
- J. Heukeshoven and R. Dernick, Electrophoresis, 1985, 6, 103–112 CAS.
- V. Neuhoff, R. Stamm and H. Eibl, Electrophoresis, 1985, 6, 427–448 CAS.
- A. Shevchenko, M. Wilm, O. Vorm and M. Mann, Anal. Chem., 1996, 68, 850–858 CrossRef CAS.
- J. X. Yan, R. Wait, T. Berkelman, R. A. Harry, J. A. Westbrook, C. H. Wheeler and M. J. Dunn, Electrophoresis, 2000, 21, 3666–3672 CrossRef CAS.
- A. Harder, R. Wildgruber, A. Nawrocki, S. J. Fey, P. M. Larsen and A. Görg, Electrophoresis, 1999, 20, 826–829 CrossRef CAS.
- A. Görg, G. Boguth, C. Obermaier, A. Posch and W. Weiss, Electrophoresis, 1995, 16, 1079–1086 CAS.
- H. Chassaigne, Detection: Electrospray Methods for Elemental Speciation, in Handbook of Elemental Speciation, ed. R. Cornelis, J. Caruso, H. Crews and K. Heumann, Wiley, Chichester, 2003, pp. 356–377 Search PubMed.
- J. R. Encinar, L. Ouerdane, W. Buchmann, J. Tortajada, R. Lobinski and J. Szpunar, Anal. Chem., 2003, 75, 3765–3774 CrossRef.
| This journal is © The Royal Society of Chemistry 2004 |