Separation of selenium compounds by CE-ICP-MS in dynamically coated capillaries applied to selenized yeast samples
Lars
Bendahl
* and
Bente
Gammelgaard
Department of Analytical Chemistry, The Danish University of Pharmaceutical Sciences, Universitetsparken 2, DK-2100 Copenhagen, Denmark. E-mail: labe@dfh.dk; Fax: +45 3530 6010
First published on 21st November 2003
Abstract
The selenium species in nutritional supplement tablets, based on selenized yeast, were separated by capillary zone electrophoresis using capillaries coated dynamically with poly(vinyl sulfonate) and detected by ICP-MS. Sample pre-treatment consisted of cold-water extraction by sonication and subsequent incubation of the cold-water extract with 6 M hydrochloric acid at 110 °C. The total selenium concentration in the cold-water extract was 3.5 mg L−1 and corresponded to 9% of the total selenium content of the tablets. More than 20 different selenium compounds were separated in the cold-water extract within 13 min. The efficiency of the system corresponded to 620![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000 theoretical plates. When spiking the sample with available standards, co-migration was observed with selenomethionine and selenocystine–Se-methylselenocysteine—the latter species were not separated. When the cold-water extract was hydrolysed in hot hydrochloric acid, 45% of the selenium migrated within a single peak that co-migrated with selenomethionine. Other peaks co-migrated with trimethylselenonium, Se-methylselenomethionine, and selenocystine–Se-methylselenocysteine, respectively. The precision for the analysis of the aqueous extracts expressed as relative standard deviation (n
= 3) on peak heights and areas was in the range 1.4–5.3%. Detection limits were better than 15 µg L−1, corresponding to absolute detection limits less than 250 fg.
000 theoretical plates. When spiking the sample with available standards, co-migration was observed with selenomethionine and selenocystine–Se-methylselenocysteine—the latter species were not separated. When the cold-water extract was hydrolysed in hot hydrochloric acid, 45% of the selenium migrated within a single peak that co-migrated with selenomethionine. Other peaks co-migrated with trimethylselenonium, Se-methylselenomethionine, and selenocystine–Se-methylselenocysteine, respectively. The precision for the analysis of the aqueous extracts expressed as relative standard deviation (n
= 3) on peak heights and areas was in the range 1.4–5.3%. Detection limits were better than 15 µg L−1, corresponding to absolute detection limits less than 250 fg.
Introduction
Speciation of selenium compounds in selenized yeast samples has recently gained increasing interest, not least due to the significant reduction in cancer incidences and mortality reported for a group of persons who had been supplemented with 200 µg Se d−1 in the form of selenized yeast.1 Selenized yeast is brewers' yeast, Saccharomyces cerevisiae, which has been grown in a selenite-enriched medium. The selenite is metabolized into organic selenium compounds, including seleno-amino acids that eventually are unspecifically incorporated into proteins. Selenized yeast contains approximately 2 mg Se g−1 and represents a common source of organic bound selenium in many nutritional supplements. Speciation of the selenium compounds in yeast preparations could provide useful information about the biochemical transformations of selenium and lead to the discovery of new selenium compounds that might have a cancer preventing effect. Additionally, speciation analysis could provide information about the quality of the selenized yeast preparation. Several studies on the speciation of selenium in selenized yeast have been performed and the results have been summarized in recent reviews.2–5HPLC is the preferred separation technique for speciation studies and more than 20 selenium compounds have been separated in aqueous yeast extracts by ion-pairing chromatography.6,7 The identities of most of the selenium compounds in yeast samples have been suggested on the basis of co-elution with authentic standards, while the presence of selenomethionine and Se-adenosyl-selenohomocysteine in yeast samples has been confirmed by electrospray ionization (tandem) mass spectrometry.8–10 Additionally, several new selenium compounds have been detected by HPLC and ESI-MS(MS), including a new class of conjugates between glutathione-S and selenium11 and some recently discovered Se-adenosyl compounds.12,13 An overview of the selenium compounds so far suggested to be present in yeast samples by HPLC-ICP-MS and ESI-MS is shown in Table 1.
| Identified species | Analytical technique | References |
|---|---|---|
| a Only applied in reference 9. | ||
| Selenomethionine | IPC-ICP-MS and (IPC-ESI-MS)a | 6,7,9,14,22,29,34–37 |
| IEC-ICP-MS | 23,38 | |
| RPC-ICP-MS | 7 | |
| Chiral LC-ICP-MS | 39 | |
| Selenocystine | IPC-ICP-MS | 6,7,34–37 |
| IEC-ICP-MS | 38 | |
| RPC-ICP-MS | 7 | |
| Se-methylselenocysteine | IPC-ICP-MS | 6,22,36,37 |
| IEC-ICP-MS | 23 | |
| Chiral LC-ICP-MS | 39 | |
| Selenite | IPC-ICP-MS | 7,22,37 |
| IEC-ICP-MS | 38 | |
| RPC-ICP-MS | 7 | |
| Se-cystathionine | IPC-ICP-MS | 37 |
| Se-lantathionine | IPC-ICP-MS | 37 |
| Chiral LC-ICP-MS | 39 | |
| Selenomethionine-Se-oxide | IEC-ICP-MS | 23 |
| Selenate | IEC-ICP-MS | 38 |
| RPC-ICP-MS and IPC-ICP-MS | 7 | |
| Selenoethionine | RPC-ICP-MS and IPC-ICP-MS | 7 |
| Trimethylselenonium | RPC-ICP-MS and IPC-ICP-MS | 7 |
| Se-adenosylselenohomocysteine | Chiral LC-ICP-MS | 39 |
| SEC/ICP-MS and off-line ESI-MS(MS) | 8 | |
| IPC-ESI-MS and IPC-ICP-MS | 9 | |
| SEC-IEC-ICP-MS and off-line ESI-MS(MS) | 12 | |
| Se-adenosyl-compounds | SEC-ICP-MS and off-line ESI-MS(MS) | 13 |
| SEC-IEC-ICP-MS and off-line ESI-MS(MS) | 12 | |
| Glutathione-S-selenium conjugates | SEC-IEC-ICP-MS and off-line ESI-MS(MS) | 11 |
The presence of selenocystine has been suggested in several studies (Table 1). Although it could be argued that selenocystine would not be present in selenized yeast as the yeast genome does not code for selenocysteine incorporation in proteins, selenocystine may be formed by other metabolic pathways. Selenoethionine is a synthetic standard and is hardly present in biological samples. On the contrary, this compound is often used as an internal standard.6,14
A large number of the separated selenium species in yeast have not yet been identified and the picture is further complicated by the fact that different fabrications of selenized yeast contain different combinations of different selenium compounds.
Capillary electrophoresis is an attractive alternative separation technique in selenium speciation due to its high separation power and its ability to separate polar and charged compounds, which are difficult to retain on reversed-phase stationary phases even when ion-paring agents are added to the mobile phase.7,14 Extracts of selenized yeast samples contain a large number of selenium compounds in high concentrations and have a low conductivity as compared with urine and plasma samples. Thus, CE-ICP-MS is in many respects a very suitable method for the analysis of yeast samples. However, concentration detection limits for selenium in CE-ICP-MS are generally two orders of magnitude higher than detection limits with HPLC-ICP-MS, and so far the technique has only been applied successfully for analysis of pre-concentrated and selenium enriched biological samples.
Only two applications of CE-ICP-MS for analysis of selenized yeast samples have been reported in the literature.10,15 Day et al. applied CE-ICP-MS for chiral speciation of L- and D-selenomethionine derivatized with 1-fluoro-2,4-dinitrophenyl-5-L-alanine amide in enzymatically digested yeast samples.15 The results supported previously reported results from chromatographic experiments in showing that only the L-form of selenomethionine was present in the samples. Recently, CE-ICP-MS was applied for analysis of the water soluble fraction of selenized yeast by Mounicou et al.10 They used an alkaline electrophoresis medium and reversed polarity detection. However, a number of the selenium compounds in the extract were not baseline separated and 25–30% of the selenium content did not leave the capillary within an analysis time of 30 min: the authors concluded that chromatographic fractionation was necessary prior to analysis by CE-ICP-MS.10 Recently, a dynamic capillary coating procedure with unique separation properties in acidic electrophoresis media was published.16
The aim of this study was to examine the suitability of capillary electrophoresis with ICP-MS detection in dynamically coated capillaries for speciation of selenium in yeast by applying the technique on yeast based selenium nutritional supplement tablets.
Experimental
CE systems and separation conditions
The CE-instrument was a Waters Quanta 4000 (Milford, MA, USA) operated with open door and defeated door interlock and separations were performed at ambient temperature. 363 µm od fused silica capillaries coated externally with poly(imide) were obtained from Poly Micro Technologies (Phoenix, AZ, USA). A 1.10 m × 50 µm id capillary was coated with poly(vinyl sulfonate) as previously described.16 The CE medium contained 100 mmol L−1 formic acid and 0.01% poly(vinyl sulfonate), pH 3.0. The CE-buffer added 10% methanol was used for sheath liquid. The run voltage was +30 kV and injection was at 9.82 mbar for 100 s (corresponding to 16.7 nL). Generally, the capillaries were flushed with the electrophoresis medium for 2 min or 5 min at 1 bar prior to analysis, and buffer reservoirs were replaced for every five or six runs.ICP-MS instrument
ICP-MS detections were performed on a PE-SCIEX ELAN 6000 instrument (PerkinElmer–Sciex Corp., Norwalk, CT, USA) configured for a demountable torch. The interface between the CE-system and the ICP-MS instrument was previously described.17 Instrumental settings are given in Table 2. Peak areas were calculated by the Turbochrom Workstation (PerkinElmer).| CE-ICP-MS | |
|---|---|
| a External argon supply. | |
| Sampling and skimmer cones | Direct injection nebulizer |
| Sample introduction system | Platinum |
| Argon flow rate | |
| Plasma gas | 1.2 L min−1 |
| Auxiliary gas | 15 L min−1 |
| Nebulisationa | 0.2 L min−1 |
| Rf power supply | 1150 W |
| Lens voltage | 6 V |
| Data acquisition | |
| Dwell time | 300 ms |
| Sweeps/reading | 1 |
| Readings/replicate | 1500 |
| Replicates | 1 |
| Isotopes monitored | 82Se |
| Sheath liquid uptake rate | 10 µL min−1 |
Selenium standards
500 µg L−1 aqueous selenium standards were prepared from selenomethionine (Sigma, St. Louis, MO, USA), selenocystine (Sigma), selenite (Titrisol 1000 mg L−1, Merck, Darmstadt, Germany) and Se-methylselenomethionine iodide synthesized by methylation of selenomethionine and trimethylselenonium iodide synthesized by methylation of dimethylselenide with methyl iodide in accordance with references 18 and 19, respectively.Extraction of selenium compounds from selenized yeast
Four nutritional supplement tablets, each containing 100 µg of selenium in the form of selenized yeast (Selenoprecise, PharmaNord, Vejle, Denmark) were sonicated in 10.0 mL of water for 1 hour at 25 °C. The resulting suspension was centrifuged at 3600g for 10 min and the pale yellow supernatant was ultra-filtered through a regenerated cellulose membrane with a cut-off at 5 kDa (Millipore Corporation, Bedford, MA, USA).2 mL of the supernatant was hydrolysed by incubation with 2 mL of concentrated hydrochloric acid in a sealed vial for 24 hours at 110 °C. The resulting solution was filtered through a 0.45 µm cellulose acetate membrane and evaporated to dryness on a rotary evaporator at 40 °C. The residue was re-dissolved in 2 mL water and re-evaporated to dryness in order to remove excess hydrochloric acid. Finally, the residue was reconstituted in 2 ml water.
The total selenium content in the extracts was determined by the method of standard additions at five concentration levels using selenite as internal standard.
Results and discussion
Extraction of selenium compounds from yeast samples
Selenized yeast contains a large number of water-soluble selenium compounds but most of the selenium is incorporated into sparingly soluble bio-molecules that are difficult to extract. Casiot et al. have evaluated eight different procedures for the extraction of selenium compounds from selenized yeast samples.20 According to their results, approximately 10% of the total selenium is released from yeast by leaching with hot water, while up to 90% of the total selenium is released when yeast samples are incubated with proteolytic enzymes. However, the latter procedure converts many of the original selenium compounds to other compounds, predominantly selenomethionine.20The selenium samples analysed in this study were nutritional supplement tablets, declared to contain 100 µg of selenium in the form of selenized yeast. In order to conserve the identity of the selenium compounds, the tablets were extracted with cold water in an ultrasonic bath followed by centrifugation and ultra-filtration of the pale yellow supernatant through a cellulose membrane with a cut-off at 5 kDa. Protective additives and buffer salts were not added as they could interfere in the stacking process in CE by increasing the conductivity of the sample, resulting in decreased resolution. The selenium concentration in the extracts was found to be 3.5 mg L−1, corresponding to 9% of the total selenium content. This is in accordance with earlier findings.20
Analysis of aqueous yeast extracts
The extracts were analysed by capillary zone electrophoresis using capillaries coated dynamically with poly(vinyl sulfonate). The inner surface of these capillaries is covered with the strongly acidic poly(vinyl sulfonate), which eliminates the influence of pH on the electroosmotic flow. Hence, the electroosmotic flow towards the cathode is maintained in the pH range 2–10.16 This is an advantage compared with the use of uncoated silica capillaries in which the weak, acidic silanol groups are protonated in acidic pH, resulting in elimination of the osmotic flow. The method was optimized with the object of separating as many selenium compounds as possible in the extract, and the best resolution was achieved when the separations were performed in relatively long capillaries using a 100 mmol L−1 formic acid electrophoresis medium, pH 3, with the addition of 0.01% poly(vinyl sulfonate). An electropherogram from the analysis of the aqueous extract is presented in Fig. 1, which shows that more than 20 different selenium compounds were separated within 13 min. The peaks were simultaneously detected by monitoring 78Se and 77Se (data not shown), supporting the view that the compounds detected were selenium compounds. However, the 82Se isotope provided the best signal to noise ratio and was monitored throughout.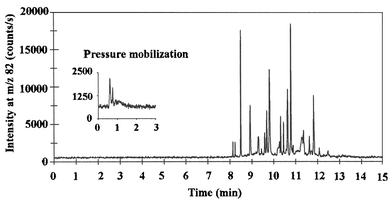 | ||
| Fig. 1 Electropherogram from analysis of the cold water extract of yeast based nutritional supplement tablets by CE-ICP-MS. Inset on the left side of the figure is a trace acquired during flushing of the capillary with 1.5 times capillary volume of buffer after the analysis was completed. Conditions: capillary, 110 cm × 50 µm id coated with poly(vinyl sulfonate); CE buffer, 100 mmol L−1 formic acid, pH 3.0, added 0.01% poly(vinyl sulfonate); sheath liquid, CE buffer added 10% methanol (10 µL min−1); run voltage, 30 kV; hydrostatic sample injection at 9.82 mbar for 100 s corresponding to 16.7 nL (60 pg Se). | ||
The efficiency of the system, calculated on the basis of the peak at a migration time of 11.85 min, was 620000 theoretical plates. This is, to our knowledge, the best separation efficiency achieved in the separation of selenium compounds by capillary electrophoresis to date. Thus, the resolution is superior to results achieved with reversed-phase, ion-pair and anion-exchange chromatography,6,7 where a column efficiency of 8500 theoretical plates in ion-pair chromatography was obtained. Moreover, the analysis time in capillary electrophoresis was one third of the analysis time in ion-pair chromatography.6
Peak widths less than 1 s at base line were achieved for some of the fastest migrating compounds. This is an order of magnitude less than the peak widths achieved in other studies.10
Inset in the left side of Fig. 1 is the trace acquired during flushing of the capillary with 1.5 times the capillary volume of buffer after the analysis was completed. The trace shows that 2 or 3 selenium species remained undetected within an analysis time of 15 min. This is in contrast to the results achieved when samples were separated in an alkaline CE buffer.10 However, this fraction only represented a small amount of the total selenium and no further effort was made to characterize these compounds.
When extracts were analyzed before the ultra-filtration through the 5 kDa membrane, no additional peaks appeared in the electropherograms; but the amount of selenium which could be flushed out with buffer after the analysis was completed increased slightly (data not shown). Thus, the majority of the selenium compounds in the water-soluble fraction were low molecular weight compounds. This is in accordance with previously reported results.6,21
Identification of selenium compounds in aqueous yeast extracts
In order to identify some of the compounds in the extract, the sample was spiked with standards of selenite, selenomethionine, selenocystine, selenoethionine, Se-methylselenomethionine, Se-methylselenocysteine, trimethylselenonium and selenocystamine. Expanded electropherograms from analysis of the extracts and extracts spiked with selenomethionine and selenocystine are presented in Fig. 2 and shows that two of the extracted compounds co-migrated with the standards. Trimethylselenonium, Se-methylselenomethionine and selenocystamine were not detected in in any of the analysed tablets and the selenite standard was not detected within 15 min. Trimethylselenonium migrated as a sharp peak with a migration time of 5.8 min and was used as an internal standard to quantify the extracted compounds. Unfortunately, selenoethionine co-migrated with selenomethionine and selenocystine co-migrated with Se-methylselenocysteine. Selenoethionine is not a common seleno-amino acid and is rarely detected in yeast samples (Table 1), hence this compound has often been used as internal standard.6,14 Thus, the unknown compound is most likely to be selenomethionine, in agreement with previously reported results listed in Table 1. Both selenocystine and Se-methylselenocysteine have been identified in yeast samples on the basis of co-elution with authentic standards. These compounds have been separated by cation-exchange, reversed phase and ion-pair chromatography6,19,22,23 and more work on optimization of the CE method with respect to separation of these seleno-amino acids is needed.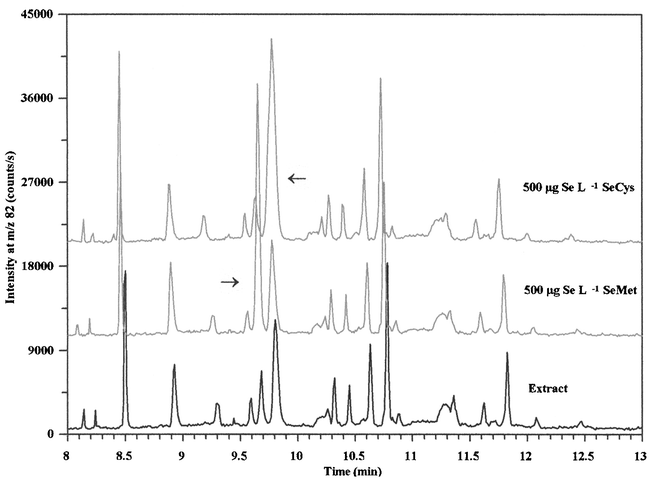 | ||
| Fig. 2 Expanded electropherograms from analysis of the cold water yeast extract by CE-ICP-MS and samples spiked with 500 µg Se L−1 selenomethionine and selenocystine, respectively. Conditions are shown in legend to Fig. 1. | ||
Semi-quantitative determinations of the selenomethionine and selenocystine/Se-methylselenocysteine concentrations in the extract showed that 8.0 ± 0.3% and 17 ± 0.5% (n = 3) of the selenium was associated with compounds that co-migrated with selenomethionine and selenocystine/Se-methylselenosysteine, respectively. This agrees with results from analysis of the same brand of nutritional supplement by reversed-phase chromatography, which showed that selenomethionine and selenocystine accounted for 13% and 14%, respectively, of the total selenium after hot water extraction.7 In addition, the same authors identified minor amounts of trimethylselenonium and selenoethionine in the extract. This difference in presence and distribution of selenium compounds in the extract could be due to batch variations and variations in sample preparation and the resolving power of the methods.
Repeatability and limits of detection
The analytical figures of merit were evaluated on the basis of the peaks which co-migrated with seleno-amino acids by analysis of three different tablets from the same batch. Data for the precision of the method with respect to migration times, peak heights and peak areas are shown in Table 3 and were better than 5.3% RSD. The precision was comparable to data reported for HPLC-ICP-MS7 and data reported for seleno-amino acid standards when analysed by CE-ICP-MS.10,24| Repeatability [RSD (%)]a (n = 3) | Detection limit | ||||
|---|---|---|---|---|---|
| Migration time | Peak height | Peak area | Relative/µg Se L−1b | Absolute/fg Sec | |
| a Precision expressed as percent relative standard deviations was calculated on the basis of results from analysis of three yeast based nutritional supplement tablets from the same batch. b The concentrations of seleno-amino acids in the extract were determined by internal standardisation with trimethylselenonium and relative detection limits were calculated as concentrations that will give signals equivalent to three times the peak-to-peak noise of the baseline. c Absolute detection limits correspond to an injected sample volume of 16.7 nL. | |||||
| Selenomethionine/selenoethionine | 0.52 | 2.9 | 3.4 | 10 | 170 |
| Selenocystine/Se-methylselenocysteine | 0.52 | 1.4 | 5.3 | 15 | 250 |
Relative detection limits expressed as the concentration that would give a signal equivalent to three times the peak-to-peak noise of the base line were better than 15 µg Se L−1, corresponding to absolute detection limits less than 250 fg of Se. The relative detection limits in this study were comparable to or better than detection limits for seleno-amino acid standards when analysed by spray chamber based CE-ICP-MS interfaces.10,24 However, relative LODs were 20 times higher than our previously reported detection limits for aqueous selenium standards in CE-ICP-MS using a direct injection nebulizer interface.17 The difference is mainly due to an eight-fold reduction of the injected sample volume. Besides, the background was high at m/z 82 when poly(vinyl sulfonate) was added to the buffer and sheath liquid, which could be due to an isobaric 34S16O3 interference.
The relative detection limits were generally 10–100 times higher with CE-ICP-MS compared with HPLC-ICP-MS due to the small sample volume injected in CE. However, many of the compounds in the aqueous yeast extracts were present in concentrations far beyond the limit of detection of CE-ICP-MS.
Absolute detection limits for CE-ICP-MS in this study were 10 times lower than data reported for HPLC-ICP-MS7,25,26 due to the high efficiency of 620000 theoretical plates with the CE-ICP-MS system.
Analysis of hydrolysed yeast extracts
The water-soluble fraction of selenized yeast has been shown to contain a large amount of low molecular selenium containing proteins by SDS-PAGE analysis.21 When yeast samples are incubated with proteolytic enzymes, significant amounts of selenomethionine are released, probably due to the hydrolysis of selenium containing proteins.In order to determine if some of the compounds detected in the cold-water extracts were seleno-amino acid containing proteins or polypeptides, the aqueous fraction was incubated with concentrated hydrochloric acid at 110 °C for 24 h. This procedure hydrolyses proteins to free amino acids and has previously been used to hydrolyse selenium containing plasma proteins27,28 and to extract selenium compounds from yeast samples.29 Electropherograms from analysis of the hydrolysate showed that 45% of the total selenium migrated within a single peak (Fig. 3). Besides, several of the compounds that were detected in the cold-water extract had disappeared and some new compounds had formed. No selenium compounds were observed when the capillary was flushed with buffer after the analysis was completed and comparisons of integration results revealed that all of the selenium in the aqueous extract was recovered after acidic hydrolysis. However, the conditions used to hydrolyse the sample were rather harsh and some of the compounds formed could be due to decomposition by other reaction paths than hydrolysis. Especially, selenocysteine is sensitive to decomposition under the conditions used for acidic hydrolysis, and is often carboxymethylated prior to hydrolysis.30–33
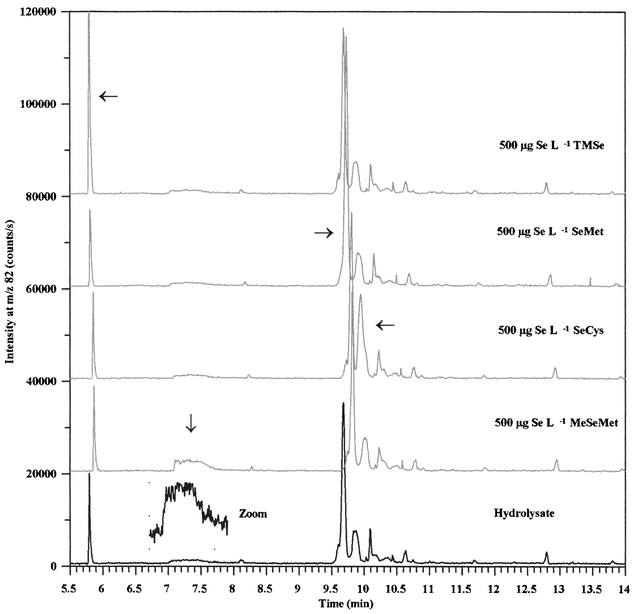 | ||
| Fig. 3 Expanded electropherograms from analysis of a hydrolysed aqueous extract of yeast and samples spiked with 500 µg Se L−1 trimethylselenonium, selenomethionine, selenocystine and Se-methylselenomethionine, respectively. Conditions are shown in the legend to Fig. 1. | ||
Identification of selenium compounds in hydrolysed yeast extracts
In order to identify some of the selenium compounds, the hydrolysate was spiked with standards as described earlier. Expanded sections of electropherograms are presented in Fig. 3, which shows that four of the compounds in the hydrolysate co-migrated with trimethylselenonium, Se-methylselenomethionine, selenomethionine and selenocystine, respectively. Semi-quantitative determinations showed that these compounds accounted for 10–45% of the total selenium in the hydrolysate. The two latter compounds were also detected in the aqueous extract but the areas of the peaks that co-migrated with selenomethionine and selenocysteine/Se-methylselenocysteine increased by a factor of 13 and 1.5, respectively, when the sample was hydrolysed. Minor amounts of Se-methylselenomethionine and trimethylselenonium have previously been identified in selenized yeast samples.7Thus, a large fraction of the selenium compounds in the cold water extracted yeast sample were predominantly converted into a compound that co-migrated with selenomethionine, suggesting that these compounds might be polypeptides or low-molecular proteins with unspecifically incorporated selenomethionyl residues.
In conclusion, we have demonstrated that capillary electrophoresis in poly(vinyl sulfonate) coated capillaries with online ICP-MS detection is a promising technique to give a finger-print of selenized yeast preparations. More than 20 selenium compounds were separated in aqueous extracts of yeast based nutritional supplement tablets within 13 min. The system had an efficiency of 620000 theoretical plates and detection limits for seleno-amino acids were in the low µg Se L−1 range.
References
- L. C. Clark, G. F. Combs, B. W. Turnbull, E. H. Slate, D. K. Chalker, J. Chow, L. S. Davis, R. A. Glower, G. F. Graham, E. G. Gross, A. Krongrad, J. L. Lesher, H. K. Park, B. B. Sanders, C. L. Smith and J. R. Taylor, JAMA, 1996, 276, 1957 Search PubMed.
- P. C. Uden, Anal. Bioanal. Chem., 2002, 373, 422 CrossRef CAS.
- R. Lobinski, J. S. Edmonds, K. T. Suzuki and P. C. Uden, Pure Appl. Chem., 2000, 72, 447 Search PubMed.
- J. Szpunar and R. Lobinski, Anal. Bioanal. Chem., 2002, 373, 404 CrossRef CAS.
- K. Pyrzynska, Microchim. Acta, 2002, 140, 55 Search PubMed.
- S. M. Bird, P. C. Uden, J. F. Tyson, E. Block and E. Denoyer, J. Anal. At. Spectrom., 1997, 12, 785 RSC.
- J. M. Marchante-Gayon, C. Thomas, I. Feldmann and N. Jakubowski, J. Anal. At. Spectrom., 2000, 15, 1093 RSC.
- C. Casiot, V. Vacchina, H. Chassaigne, J. Szpunar, M. Potin-Gautier and R. Lobinski, Anal. Commun., 1999, 36, 77 RSC.
- M. Kotrebai, M. Birringer, J. Tyson, E. Block and P. C. Uden, Anal. Commun., 1999, 36, 249 RSC.
- S. Mounicou, S. McSheehy, J. Szpunar, M. Potin-Gautier and R. Lobinski, J. Anal. At. Spectrom., 2002, 17, 15 RSC.
- S. McSheehy, P. Pohl, J. Szpunar, M. Potin-Gautier and R. Lobinski, J. Anal. At. Spectrom., 2001, 16, 68 RSC.
- S. McSheehy, F. Pannier, J. Szpunar, M. Potin-Gautier and R. Lobinski, Analyst, 2002, 127, 223 RSC.
- S. McSheehy, J. Szpunar, V. Haldys and J. Tortajada, J. Anal. At. Spectrom., 2002, 17, 507 RSC.
- P. C. Uden, S. M. Bird, M. Kotrebay, P. Nolibos, J. F. Tyson, E. Block and E. Denoyer, Fresenius’ J. Anal. Chem., 1998, 362, 447 CrossRef CAS.
- J. A. Day, S. S. Kannamkumarath, E. G. Yanes, M. Montes-Bayón and J. A. Caruso, J. Anal. At. Spectrom., 2002, 17, 27 RSC.
- L. Bendahl, S. H. Hansen and B. Gammelgaard, Electrophoresis, 2001, 22, 2565 CrossRef CAS.
- L. Bendahl, B. Gammelgaard, O. Jøns, O. Farver and S. H. Hansen, J. Anal. At. Spectrom., 2001, 16, 38 RSC.
- S. J. Foster and H. E. Ganther, Anal. Biochem., 1984, 137, 205 CrossRef CAS.
- T. W. M. Fan, A. N. Lane, D. Martens and R. M. Higashi, Analyst, 1998, 123, 875 RSC.
- C. Casiot, J. Szpunar, R. Lobinski and M. Potin-Gautier, J. Anal. At. Spectrom., 1999, 14, 645 RSC.
- C. C. Chéry, H. Chassaigne, R. Verbeeck, R. Cornelis, F. Vanhaeke and L. Moens, J. Anal. At. Spectrom., 2002, 17, 576 RSC.
- M. Kotrebai, S. M. Bird, J. F. Tyson, E. Block and P. C. Uden, Spectrochim. Acta, Part B, 1999, 54, 1573 CrossRef.
- E. H. Larsen, M. Hansen, T. Fan and M. Vahl, J. Anal. At. Spectrom., 2001, 16, 1403 RSC.
- B. Michalke, LC-GC Europe, 2000,(January), 36 Search PubMed.
- I. Feldmann, N. Jakubowski, C. Thomas and D. Stuewer, Fresenius’ J. Anal. Chem., 2003, 365, 422 CrossRef.
- I. Feldmann, N. Jakubowski, D. Stuewer and C. Thomas, J. Anal. At. Spectrom., 2000, 15, 371 RSC.
- Y. Saito, T. Hayashi, A. Tanaka, Y. Watanabe, M. Suzuki, E. Saito and K. Takahashi, J. Biol. Chem., 1999, 274, 2866 CrossRef CAS.
- J. T. Deagan, M. A. Beilstein and P. D. Whanger, J. Inorg. Biochem., 1991, 41, 261 CrossRef CAS.
- C. B'Hymer and J. A. Caruso, J. Anal. At. Spectrom., 2000, 15, 1531 RSC.
- K. E. Hill, R. S. Lloyd, J. Yang, R. Read and R. F. Burk, J. Biol. Chem., 1991, 266, 10050 CAS.
- R. Read, T. Bellew, J. Yang, K. E. Hill, I. S. Palmer and R. F. Burk, J. Biol. Chem., 1990, 265, 17899 CAS.
- R. Daher and F. V. Lente, Clin. Chem., 1994, 40, 62.
- C. Hammel, A. Kyriakopoulos, U. Rösick and D. Behne, Analyst, 1997, 122, 1359 RSC.
- R. M. Olivas, O. F. X. Donard, N. Gilon and M. Potin-Gautier, J. Anal. At. Spectrom., 1996, 11, 1171 RSC.
- J. Zheng, M. Ohata, N. Furuta and W. Kosmus, J. Chromatogr. A, 2000, 874, 55 CrossRef CAS.
- S. M. Bird, H. G. Ge, P. C. Uden, J. F. Tyson, E. Block and E. Denoyer, J. Chromatogr. A, 1997, 789, 349 CrossRef CAS.
- M. Kotrebai, M. Birringer, J. F. Tyson, E. Block and P. C. Uden, Analyst, 2000, 125, 71 RSC.
- J. Zheng, W. Goessler and W. Kosmus, Trace Elem. Electrolytes, 1998, 15, 70 Search PubMed.
- M. Kotrebai, J. F. Tyson, E. Block and P. C. Uden, J. Chromatogr. A, 2000, 866, 51 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2004 |