Selenium speciation in human urine samples by LC- and CE-ICP-MS—separation and identification of selenosugars
Bente Gammelgaard* and Lars Bendahl
Department of Analytical Chemistry, The Danish University of Pharmaceutical Sciences, Universitetsparken 2, DK-2100 Copenhagen, Denmark. E-mail: bg@dfh.dk; Fax: +45 3530 6010
First published on 26th September 2003
Abstract
Human urine samples were analysed by a reversed-phase chromatographic system and an ion-pair chromatographic system. The chromatographic system was connected to the ICP-MS either by a microconcentric nebulizer (MCN) in combination with a cyclonic spraychamber or by a modified direct injection nebulizer (MDIN). The sensitivity of the latter was better than the sensitivity of the MCN, which on the other hand was more robust for the analysis of samples with high concentrations of dissolved solids. Urine sample composition did not seem to change when urine was exposed to evaporation under nitrogen at ambient temperature and methanol extraction. A pre-concentration factor of 10 was achieved with this procedure. On occasions when a pre-concentration factor of 100 was obtained by lyophilsation and methanol extraction, at least 10 selenium compounds were separated in the urine sample. Urine samples were collected from two healthy volunteers who had been supplied with 1000 µg and 2000 µg of selenium, respectively, in the form of selenized yeast. When samples were spiked with 8 different standards, only two standards co-eluted with compounds in urine in both chromatographic systems: the major urinary metabolite Se-methyl-N-acetylgalactosamine and Se-methyl-N-acetylglucosamine. The presence of Se-methyl-N-acetylglucosamine in urine was verified by co-migration with the standard in capillary electrophoresis after fractionation by preparative reversed-phase chromatography. Se-methyl-N-acetylglucosamine is only a minor metabolite as its concentration was less than 2% of the concentration of Se-methyl-N-acetylgalactosamine. The presence of this metabolite in urine has, to our knowledge, not been suggested before. Trimethylselenonium, selenomethionine, Se-methylselenocysteine, Se-methylselenomethionine and selenocystamine were not detected in these samples.
Introduction
The essential trace mineral selenium is indispensable for the functioning of the human body. Selenium exerts its effects via the selenoproteins that often participate in anti-oxidative or catalytic processes. A review on selenium-containing natural products, including selenoproteins, has recently been given.1 According to this, 18 mammalian selenoproteins have now been defined by sequence.Selenium deficiency causes various adverse health effects2 and in recent years the element has gained increasing interest owing to its possible cancer protective effects.3 Results of experiments on the cancer protective effect of monomethylated selenium compounds such as selenobetaine, Se-methylselenocysteine and methylseleninic acid suggest that formation of a monomethylated selenium metabolite is important for cancer chemoprevention.4–6
Selenium metabolism
The most often presented model on the metabolism of selenium is primarily based on experiments on rats after administration of large doses of selenium.7,8 According to this model, selenomethionine is either incorporated unspecifically into proteins in competition with methionine or is transformed via selenocysteine to selenide, from which it enters the selenoproteins after synthesis to selenocysteine. Excess selenium is excreted via methylation to monomethyl selenol, dimethylselenide, which is exhaled via the breath, and to trimethylselenonium which is excreted in urine. This pathway is shown in Fig. 1 as the pathway for toxic levels.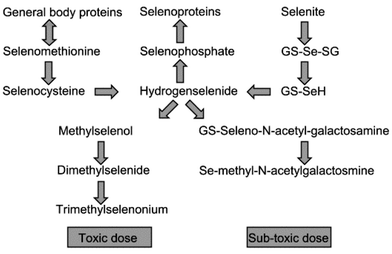 | ||
| Fig. 1 Model of selenium metabolism modified from the original model by Ip5 and supplemented with the latest results of the work from the group of Suzuki.13 GS-: Glutathion conjugate. | ||
The metabolism of inorganic selenium in sub-toxic doses in rats has been thoroughly described by Suzuki and co-workers on the basis of administration of 82Se-enriched selenite and selenate and subsequent determination of the 82Se level in organs and body fluids by size exclusion chromatography and ICP-MS detection.9 Intravenously injected selenite was selectively taken up by the red blood cells (RBC)10 and reduced by glutathion (GSH) to selenide. Selenide was transported to the liver, where it was incorporated into selenoprotein P, re-excreted to the bloodstream and transferred to the kidneys, where it was degraded and used for synthesis of extra-cellular glutathion peroxidase.9 Contrary to selenite, selenate was not taken up by RBCs but was retained in the bloodstream until it entered the liver or was excreted directly in urine. The metabolic products of selenite and selenate were not distinguishable.11 The selenium in liver was distributed between two compounds, the major urinary metabolite Se-methyl-N-acetylgalactosamine12 and a compound in which the methyl group on selenium was exchanged with glutathione. It was suggested that this compound was a precursor of the major urinary metabolite.13 This pathway is shown in Fig. 1 as the pathway for non-toxic doses. The metabolism of selenium in humans is presumed to follow the same ways.
Identification of selenium compounds in human urine
Only few data on human selenium metabolites are available. With the introduction of ICP-MS, speciation in human urine was made possible owing to the high sensitivity of this technique. An overview of the metabolites identified in human urine by on-line separation-ICP-MS detection is shown in Table 1.| Selenium compound | Analytical techniquea | Reference |
|---|---|---|
| a IPC: Ion-pair chromatography; RPC: reversed-phase chromatography; CEC: cation exchange chromatography; AEC: anion exchange chromatography; MS: mass spectrometry; HG: hydride generation; MIP: microwave induced plasma. | ||
| Trimethylselenonium | IPC-ICP-MS | 14–16 |
| RPC-ICP-MS | 16–18 | |
| CEC-ICP-MS | 19,20 | |
| Selenite | IPC-ICP-MS | 14 |
| RPC-ICP-MS | 21 | |
| AEC-ICP-MS | 22 | |
| AEC-HG-N2-MIP-MS | 23 | |
| Selenomethionine | CEC-ICP-MS, CE-ICP-MS | 19 |
| RPC-ICP-MS, tandem MS/MS | 24 | |
| IPC-ICP-MS | 18 | |
| Selenocystamine | RPC-ICP-MS, tandem MS/MS | 24 |
| Se-methyl-N-acetylgalactosamine | RPC-ICP-MS, MS/MS | 25 |
In most of the studies, the identities of the selenium compounds were suggested on the basis of co-elution with standards. However, recently some metabolites have been identified by electrospray ionisation mass spectrometry (ESI-MS). Selenomethionine and selenocystamine were identified by tandem mass spectrometry in urine from an adult male who had been supplemented with a single dose of 400 µg of selenomethionine. The multiple reaction monitoring mode, in which one or more transitions of precursor to product ions are monitored in tandem, was used for the identification.24 The major metabolite in human urine was identified by atmospheric pressure chemical ionisation mass spectrometry (APCI-MS) in a human urine pool obtained from 6 male volunteers supplemented with 200 µg of selenium-enriched yeast a day for 1 week. This metabolite was Se-methyl-N-acetylgalactosamine25—the same metabolite as the one previously obtained from rats after supplementation with selenite.12
Apart from the selenium compounds listed in Table 1, the presence of selenocholine and selenourea in urine has been cited in reviews. However, going into the original references, it appears that standards of these species may have been separated in a urine matrix, but they were not identified in the original urine samples.26
Sample preparation
In the most efficient separation systems, more than 5 different selenium compounds were separated in human urine.15,16,18,19,24,25 Hence, several metabolites in human urine still remain to be identified. Supplementation with selenium, however, does not increase the concentrations of these compounds in appreciable amounts compared with the major metabolite. Hence, identification of these compounds by ESI-MS demands intensive pre-concentration procedures as the sensitivity of ESI-MS in the full scan mode is about two orders of magnitude poorer than the sensitivity for ICP-MS. However, if the identity of an unknown compound was indicated by co-elution with a standard in LC-ICP-MS, the ESI-MS instrument could be optimised on the expected m/z value and this would increase the sensitivity appreciably.Most of the identifications represented in Table 1 were performed on untreated samples that were only diluted and or filtrated.14–16,20–22,27,28 Others used solid-phase extraction on C18 cartridges to remove uncharged organic compounds,17 but also more aggressive procedures as crown ether extraction has been applied to remove potassium and sodium from the samples.19
Pre-concentration prior to MS-identification includes lyophilisation followed by chromatographic fractionation,24 incubation with urease at 45 °C followed by evaporation and extraction with cold methanol, prior to chromatographic fractionation,12 and solid phase extraction, followed by chromatographic fractionation on reversed phase and size exclusion columns.25 In a previous study we observed that the major metabolite decomposed into another compound within 24 h at ambient temperature,18 hence it is crucial for identification studies that the analytical procedures preserve the original selenium compounds.
The aim of this study was to develop analytical procedures for separation of selenium compounds in human urine and to investigate if the sample preparation procedures usually applied for pre-concentration of samples change the sample composition. A secondary aim was to identify the compounds in urine by co-elution with available standards with the future aim of identifying new compounds by ESI- or APCI-MS.
Experimental
Apparatus
The flow rate was 200 µl min−1 and the sample injection volume was 12 µl when 2 mm id columns were used. When 1 mm id columns were used the flow rate was 50 µl min−1 and the sample injection volume was 3 µl.
Reversed phase chromatography. The columns were two Luna C18(2), 100 mm × 2 mm id, 3 µm particle size, in series (Phenomenex, Aschaltenburg, Germany). The eluent was 200 mM ammonium acetate + 5% methanol.
Ion-pair chromatography. The column was a Luna C8(2), 100 mm × 2 mm id, or a 100 mm × 1 mm id, 3 µm particle size (Phenomenex). The eluent was 0.2% heptafluorobutanoic acid (HFBA) + 20% or 30% methanol.
The chromatographic system was coupled to the ICP-MS system via 175 µm id PEEK tubing (Upchurch Scientific, Oak Harbor, WA, USA).
Buffer: 25 mM borate diluted from “buffer pH 9.3 for HPCE” (Agilent Technologies); sheath liquid: 5% acetic acid in 0.67% nitric acid, flow rate 10 µl min−1.
The ICP-MS was a PE SCIEX ELAN 6000 (PerkinElmer, Norwalk, CT, USA equipped with a MicroMist AR30-1-F02 glass concentric nebulizer and a cyclonic spray chamber (Glass Expansion, Romainmotier, Switzerland). The spray chamber was cooled to −4 °C when the eluent contained more than 5% methanol. A direct injection nebulizer that has previously been described29 was used for interfacing CE and 1 mm id columns.
Sampler and skimmer cones were of platinum. The plasma and auxiliary argon flow rates were 14 and 1.2 L min−1, respectively. The nebulization argon gas flow rate was 1.025 L min−1 for the microconcentric nebulizer and 0.2 L min−1 for the direct injection nebulizer. The rf power was 1300 W (CE) and 1425 W.
The data acquisition parameters were: dwell time, 300 ms; sweeps per reading, 1; readings per replicate, 750/1500 (CE).
The 77Se, 78Se and 82Se isotopes were monitored. To achieve maximum sensitivity, the instrument was optimised with respect to the nebulizer gas flow rate, ion lens voltage and rf power when aspirating each eluent spiked with selenite.
Peak heights and peak areas were calculated by Turbochrom (PerkinElmer).
Stock standard solutions of 10 mg Se L−1 were prepared in water from selenomethionine (Sigma), Se-methylselenocysteine (Sigma), selenocystamine (Sigma), selenoethionine (Sigma), trimethylselenonium iodide (synthesized according to ref. 30), Se-methylselenomethionine (synthesized according to ref. 31), Se-methyl-N-acetylglucosamine (synthesized in a modified version according to ref. 12) and Se-methyl-N-acetylgalactosamine (synthesized at the Department of Medical Chemistry, The Danish University of Pharmaceutical Sciences). The stock standard solutions were standardized against a 1.001 g Se L−1 PE pure atomic spectroscopy standard (PerkinElmer). Working standards were prepared daily in water.
A: 1000 µl of urine was evaporated in a stream of nitrogen at ambient temperature and reconstituted in 100 µl of eluent.
B: 1000 µl of urine was evaporated in a stream of nitrogen at ambient temperature and extracted with 1000 µl of methanol in an ultrasonic bath for 5 min. The extract was centrifuged at 14![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000 rpm for 5 min and the supernatant evaporated to dryness at ambient temperature and reconstituted in 100 µl of eluent.
000 rpm for 5 min and the supernatant evaporated to dryness at ambient temperature and reconstituted in 100 µl of eluent.
The sediment from the centrifugation was reconstituted in 100 µl of eluent and analysed.
C: 100 ml of urine was lyophilised and extracted with 10 ml of methanol by sonication for 5 min. The extract was centrifuged 5300 rpm for 5 min and the supernatant was evaporated to dryness in a stream of nitrogen at ambient temperature and reconstituted in 1 ml of eluent.
Urine samples analysed in the HFBA system were adjusted to pH 2 by addition of concentrated HFBA.
Turbid samples were filtered through a 0.45 µm cellulose membrane (Cromacol Ltd., Trumbull, CT, USA) prior to analysis.
Results and dicussion
Two different interfaces were compared: a modified direct injection nebulizer (MDIN) and a microconcentric nebulizer (MCN) in combination with a cyclonic spraychamber. The MDIN was used in combination with chromatographic columns with inner diameters of 1 mm and a flow rate of 50 µl min−1. The MCN has an optimum flow rate at 200 µl min−1 and was used in combination with chromatographic columns with inner diameters of 2 mm.The 77Se, 78Se and 82Se isotopes were monitored simultaneously. The S/N ratio for 78Se was about half the value of the S/N ratio for 77Se and 82Se when using the MCN, so 82Se was chosen for this system. Using the MDIN, the S/N ratio was better for 78Se than for 77Se and 82Se and this isotope was chosen for the direct injection system.
Chromatographic systems—selenium standards
Two different chromatographic systems were used: a reversed phase system based on 200 mM ammonium acetate + 5% methanol and an ion-pair system based on 0.2% HFBA + 20% (or 30%) methanol. The concentration of ammonium acetate in the reversed phase system was optimised regarding resolution and peak shapes. Increasing the concentration to the relatively high concentration of 200 mM improved the peak shapes of some of the standards, resulting in almost symmetrical and well separated peaks.Chromatograms of selections of the 8 aqueous selenium standards trimethylselenonium (TMSe), Se-methylselenocysteine (MeSeCys), Se-methylselenomethionine (MeSeMet), selenocystamine (SeCA), selenomethionine (SeMet), Se-methyl-N-acetylgalactosamine (SeGal), Se-methyl-N-acetylglucosamine (SeGlu) and selenoethionine (SeEt), each in a concentration of 10 µg L−1, in the different chromatographic systems coupled via the different interfaces, are shown in Fig. 2. It appears that the signal to noise ratio of the MDIN was superior to the MCN in combination with the cyclonic spraychamber. Furthermore, the monitoring of 78Se with the more favourable signal to noise ratio using the direct injection nebulizer further improved the sensitivity as the abundance of this isotope is 23.8% compared with 8.7% for 82Se. The detection limits calculated as the concentration that would give a signal equivalent to 3 times the peak-to-peak noise at the baseline in each system are given in Table 2. These detection limits were calculated for different nebulizers in combination with columns with different inner diameters operated at different flow rates. Hence, the detection limits represent the total chromatographic system including the interface. These detection limits are in accordance with previously reported values, the majority being below 2 µg L−1.14–18,20–23,27,28
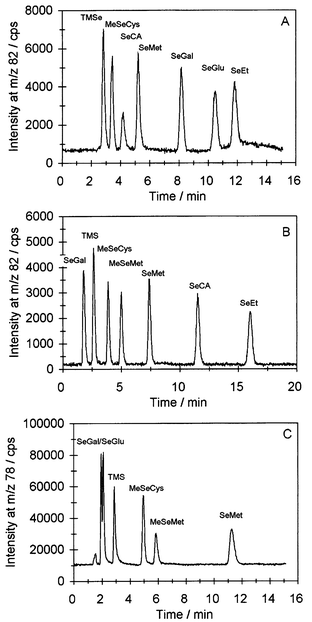 | ||
| Fig. 2 Chromatograms of aqueous standards in concentrations of 10 µg Se L−1 each. A, Reversed-phase chromatography, eluent 200 mM ammonium acetate + 5% methanol, nebulizer MCN. B, Ion-pair chromatography, eluent 0.2% HFBA + 30% methanol, nebulizer MCN. C, Ion-pair chromatography, eluent 0.2% HFBA + 20% methanol, nebulizer MDIN. | ||
When large numbers of concentrated urine samples were analysed, the micro-bore column used in combination with the MDIN quickly deteriorated and the tip of the nebulizer often clogged due to salt crystals. Hence, the MCN in combination with the 2 mm inner diameter columns was chosen for some applications in defiance of its lower sensitivity, as it was more robust for analysis of large numbers of concentrated urine samples.
None of the chromatographic systems were capable of separating all the available standards in a reasonable time. Only in the reversed phase system were the selenosugars SeGal and SeGlu separated satisfactorily. However, in this system TMSe, and other charged compounds, were not retained. The HFBA system retained TMSe, while the uncharged selenosugars were only weakly retained and hardly separated. In the HFBA system containing 20% methanol, the retention times of SeCA and SeEt were 22 min and 33 min, respectively, and resulted in intensive peak broadening. Hence, 30% methanol was used when these compounds were analysed.
Some of the standards were chosen as they have previously been identified in the human urine samples: TMSe, SeCA, SeMet and SeGal (Table 1). SeGlu was chosen as in our previous experiments with fractionation and pre-concentration of urine for APCI-MS identification of SeGal, a compound which co-eluted with SeGlu appeared. Hence, it would be interesting if this compound could be identified in urine. SeEt was included as a possible internal standard.
Urine samples—sample preparation
Chromatograms of an undiluted fresh urine sample superimposed with the same sample after spiking with selected standards in the reversed phase chromatographic systems are shown in Fig. 3A. The same sample analysed after evaporation under nitrogen, followed by reconstitution in eluent (procedure A) is shown in Fig. 3B. The sample was concentrated 10 times with this procedure and some of the compounds from the untreated sample became more visible, but no additional peaks were observed, which would indicate that the compounds in the sample had changed. By extracting the evaporated sample with methanol followed by centrifugation, evaporation of methanol, and reconstitution in eluent (procedure B), the same number of compounds were observed, Fig. 3C. No difference in the resolution was observed: the chromatograms in Fig. 3B and 3C were almost identical. Thus, methanol extraction did not change the sample. This additional procedure diminished the salt concentration in the sample, which could be beneficial prior to CE-analysis, which is very difficult in samples with high salt contents. Also a small improvement in sensitivity was obtained near the void volume in the chromatogram.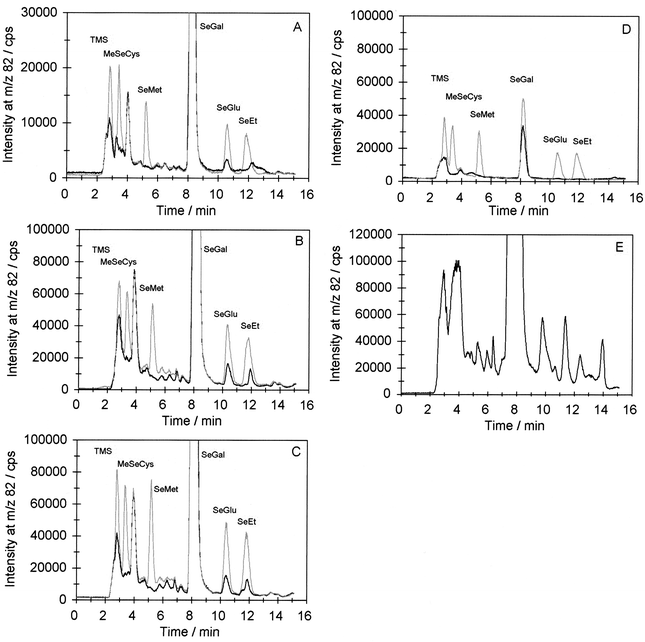 | ||
| Fig. 3 Chromatograms of urine samples after different sample pre-concentration procedures. Reversed phase chromatography: eluent 200 mM ammonium acetate + 5% methanol, nebulizer MCN. A: Undiluted urine sample, black line; sample spiked with standards in concentrations of 20 µg Se L−1 each, grey line. B: Evaporated urine sample (pre-concentrated 10 times), black line; sample spiked with standards in concentrations of 100 µg Se L−1 each, grey line. C: Methanol extracted urine sample (pre-concentrated 10 times), black line; sample spiked with standards in concentrations of 100 µg Se L−1 each, grey line. D: Extraction sediment, black line; sediment spiked with standards in concentrations of 100 µg Se L−1 each, grey line. E: Lyophilised urine sample (pre-concentrated 100 times). | ||
Fig. 3D shows an analysis of the sediment from the centrifugation in the methanol extractions. It appears that the majority of the selenium was extracted with methanol: the total loss of SeGal in the extraction was less than 5%.
Fig. 3E shows a volume of 100 ml of the same urine that was lyophilised, methanol extracted and reconstituted in 1 ml of eluent, corresponding to a concentration factor of 100. It appears that about 10 selenium compounds were more or less separated. Spiking with standards only showed co-elution with SeGal and SeGlu and the chromatography was poor as this sample contained approximately 50% dissolved solids. Hence, pre-concentration in this scale is only suitable and necessary for fractionation of samples by preparative chromatography prior to MS identification. However, the shapes of the chromatograms from the different pre-concentration procedures look very similar, which indicates that the selenium compounds in the original undiluted sample were not changed noticeably by the gentle procedures of evaporation under nitrogen at ambient temperature, lyophilisation, or methanol extraction. Hence, these procedures could be used for pre-concentration of samples prior to identification.
Identification of selenium compounds in urine
When urine samples were spiked with selected standards in the reversed phase system, co-elution with SeGal and SeGlu was observed. SeMet and SeEt did not co-elute with compounds in these urine samples, while TMSe and MeSeCys eluted close to the void volume where the resolution is poor, hence it is questionable if these standards co-eluted with compounds in the urine, Fig. 3A. This picture was even more clear in the methanol extract in Fig. 3C, except that a compound in the extract now seemed to co-elute with SeEt.When the samples were analysed in the ion-pair system, the absence of TMSe and MeSeCys was confirmed in the undiluted as well as the methanol extracted sample. This is shown in Fig. 4A and 4B. Thus, only SeGal and SeGlu co-eluted with compounds in urine in both systems.
 | ||
| Fig. 4 Chromatograms of urine samples. Ion-pair chromatography, eluent 0.2% HFBA + 20% methanol, nebulizer MDIN. A: Undiluted urine sample, black line; sample spiked with standards in concentrations of 50 µg Se L−1 each, grey line. B: Methanol extracted urine sample, black line; sample spiked with standards in concentrations of 100 µg Se L−1 each, grey line. | ||
One unidentified compound with a retention time of 4 min was observed in the reversed phase system. The retention times of aqueous standards were almost identical to the retention times in spiked urine samples. The urine sample in Fig. 3A and B was spiked with SeCA, but no increase in peak size was observed at the retention time of SeCA. It was later observed that adding SeCA to urine samples prior to analysis in the reversed phase system resulted in an indefinable peak.
To examine if SeEt or SeCA were present in urine, the undiluted urine sample, as well as the methanol extract, was analysed in an ion-pair system containing 30% methanol, which was capable of separating aqueous standards of these compounds, Fig. 2B. No co-elution was observed with urine compounds in this system (data not shown). Hence, the presence of SeEt and SeCA in this sample was excluded.
CE separation of SeGal and SeGlu
As the selenosugars were only separated in the reversed phase system, a CE system was developed to verify the presence of SeGlu in urine. The separation was based on the fact that sugars form charged complexes with borate. The sample was pre-concentrated 10 times by evaporation in a stream of nitrogen and applied to a preparative reversed phase column. A chromatogram of the separation is shown in Fig. 5A, superimposed with a chromatogram of aqueous SeGal and SeGlu standards. The fractions in the shaded area were collected and further concentrated; the final concentration factor was 200. Separation of the selenosugar standards in concentrations of 1 mg Se L−1 is shown in Fig. 5B; it appears that well shaped, narrow peaks and baseline separation were achieved. The electropherogram of the concentrated sample is shown in Fig. 5C. Owing to insufficient removal of ammonium acetate in the sample, this peak is broader than the peaks of the aqueous standards, resulting in some sensitivity suppression. The electropherogram in Fig. 5D shows the sample after addition of the selenosugar standards. It appears that the seleno-compound in the sample co-migrated with SeGlu. As the selenium compound co-eluted and co-migrated, respectively, with SeGlu in two orthogonal systems, it seems probable that SeGlu is present in the sample.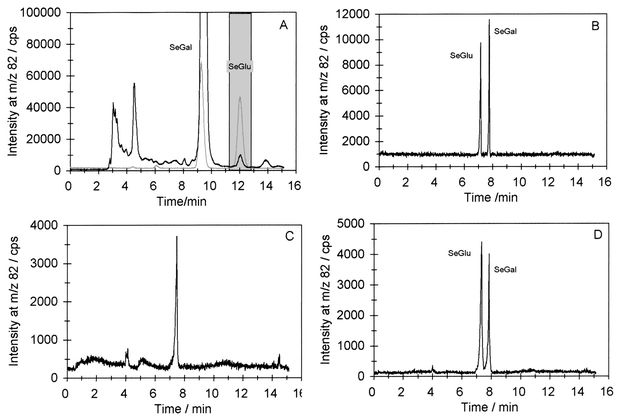 | ||
| Fig. 5 Identification of SeGlu. A: Preparative chromatography of pre-concentrated sample, black line; superimposed with 100 µg Se L−1 standard addition of SeGal and SeGlu, grey line; fractions collected, shaded area. B. Electropherogram of SeGal and SeGlu standards in concentrations of 1 mg Se L−1. C: Electropherogram of collected fractions from A. D: Collected fractions spiked with 1 mg Se L−1 SeGal and SeGlu. | ||
The presence of SeGlu in urine has, to our knowledge, not been reported before. This could be due to the lack of commercially available standards or due to the very low concentration of this compound in urine. SeGlu is only a minor metabolite: the concentration in this sample was less than 2% of the SeGal concentration and the metabolite was only detectable after consumption of relatively large amounts of selenium.
Stability of compounds
Two problems arise with sample preparation for the identification of selenium compounds in urine: the original compounds may be lost during sample preparation or they may be transformed into new compounds that may erroneously be identified as original compounds.From our previous experiments it was known that SeGal was partly decomposed in 0.1% HFBA within 24 h, probably owing to the acidic pH adjustment of the samples with the purpose of converting amino groups in selenium compounds to cationic species prior to analysis in the anionic ion-pair medium.18 Hence, this chromatographic system is not suitable for long series of analyses as the samples decompose in the autosampler. Also decomposition of untreated urine samples was observed in this study. Fig. 6 shows chromatograms of a urine sample kept at ambient temperature for 3 d. It appears from both chromatograms that new unidentified selenium containing compounds with a retention time of about 6.5 min in the reversed phase system, Fig. 6A, and 11 min in the ion-pair system, Fig. 6B, were formed from degradation of SeGal.
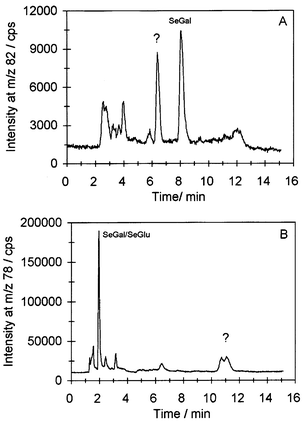 | ||
| Fig. 6 Chromatograms of urine sample after storage for 3 d at ambient temperature. A: Eluent 200 mM ammonium acetate + 5% methanol, nebulizer MCN. B: Eluent 0.2% HFBA + 20% methanol, nebulizer MDIN. | ||
When standards were added to urine and analysed in the reversed phase system both immediately and after 3 days it appeared that TMSe, MeSeCys, MeSeMet and SeEt were stable in urine samples for this time span at ambient temperature. It was rather surprising that MeSeMet was stable in urine as this compound is very unstable in aqueous solution. The concentrations of SeGal, SeGlu and SeMet were more or less halved. SeCA showed an indefinable peak from the beginning. These findings are in accordance with findings by others. Loss of SeMet in urine stored at 25 °C, together with appearance of new unidentified compounds, have been reported.32,33 Also, immediate decomposition of SeCA after its addition to urine has been reported.15
To determine whether the selenium compounds would be recovered from the method of freeze drying combined with methanol extraction in preparation for identification with MS, urine samples were spiked with selected standards and extracted. TMSe, SeMet, SeGal and SeGlu were all extracted into methanol. SeCA was also added to the sample but the peak with a retention time of 4 min that co-eluted with SeCA (Figs. 2A and 3A) did not increase. Hence, the unidentified compound was extracted and SeCA was not.
These results show that the identities of the compounds have to be known before the stability can be examined. Regarding quantitative determinations, further stability studies and recovery experiments at different storage temperatures should be performed on the relevant standards. These are initiated in our laboratory.
Application on 77Se enriched urine samples
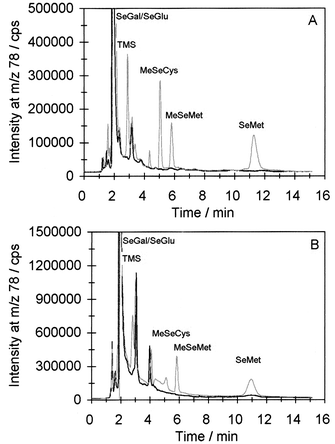
The reversed phase system was applied to a few urine samples from people who had received a single dose of 327 µg of 77Se in the form of 77Se-labelled yeast after supplementation with 300 µg of selenium per day as selenized yeast for six weeks. A chromatogram of urine samples from the day before and from the day of 77Se consumption is shown in Fig. 7A. The full-sized SeGal peak corresponded to 400000 cps. Comparing the 77Se traces before and after 77Se consumption shows that the concentration of most of the selenium species increased within the 24 h. This suggests that the metabolites in urine are formed very quickly and a large part of it is immediately excreted, predominantly as SeGal. This also shows the advantage of using enriched isotopes as the sensitivity is increased compared with monitoring selenium isotopes in samples with the natural abundance distribution. Fig. 7B shows a spiked urine sample from another person in the same study.34 It appears that also this sample shows co-elution with SeGlu.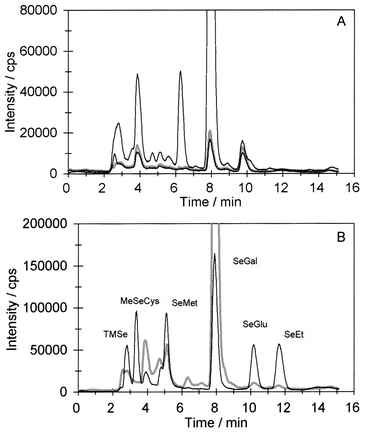 | ||
| Fig. 7 Chromatograms of methanol extracted urine samples collected before and after ingestion of 77Se-labelled yeast. A: 77Se before ingestion of 77Se, grey line; 82Se before ingestion of 77Se-labelled yeast, lower black line; 77Se after ingestion of 77Se-labelled yeast, upper black line (full-size peak height of SeGal, 400000 cps). B: Urine sample spiked with selected standards. 77Se after ingestion of 77Se-labelled yeast, grey line; 82Se after spiking with standards in concentrations of 100 µg Se L−1, black line. | ||
In conclusion, various separation systems can be combined to identify selenium compounds in urine as one system rarely has the resolving power to separate all compounds. Evaporation of urine samples at ambient temperature, lyophilisation and methanol extraction do not change urine samples and can be used to pre-concentrate samples for identification. Two metabolites were identified in the urine samples, the major metabolite Se-methyl-N-acetylgalactosamin and a novel metabolite attributed to Se-methyl-N-acetylglucosamine by orthogonal separation techniques.
References
- M. Birringer, S. Pilawa and L. Flohé, Nat. Prod. Rep., 2002, 19, 693 RSC.
- M. P. Rayman, Proc. Nutr. Soc., 2002, 61, 203 Search PubMed.
- L. C. Clark, G. F. Combs, B. W. Turnbull, E. H. Slate, D. K. Chalker, J. Chow, L. S. Davis, R. A. Glover, G. F. Graham, E. G. Gross, A. Krongrad, J. L. Lesher, H. K. Park, B. B. Sanders, C. L. Smith and J. R. Taylor, JAMA, 1996, 276, 1957 Search PubMed.
- H. E. Ganther and J. R. Lawrence, Tetrahedron, 1997, 53, 12299 CrossRef CAS.
- C. Ip, J. Nutr., 1998, 128, 1845 CAS.
- C. Ip, H. J. Thompson, Z. Zhu and H. E. Ganther, Cancer Res., 2000, 60, 2882 CAS.
- H. E. Ganther, J. Am. Coll. Toxicol., 1986, 5, 1 Search PubMed.
- S. J. Foster, R. J. Kraus and H. E. Ganther, Arch. Biochem. Biophys., 1986, 247, 12 CrossRef CAS.
- K. T. Suzuki and Y. Ogra, Food Addit. Contam., 2002, 19, 974 CrossRef CAS.
- Y. Shiobara and K. T. Suzuki, J. Chromatogr. B, 1998, 710, 49 CrossRef CAS.
- Y. Kobayashi, Y. Ogra and K. T. Suzuki, J. Chromatogr. B, 2001, 760, 73 CrossRef CAS.
- Y. Ogra, K. Ishiwata, H. Takayama, N. Aimi and K. T. Suzuki, J. Chromatogr. B, 2002, 767, 301 CrossRef CAS.
- Y. Kobayashi, Y. Ogra, K. Ishiwata, H. Takayama, N. Aimi and K. T. Suzuki, PNAS, 2002, 99, 15932 Search PubMed.
- K. Yang and S. Jiang, Anal. Chim. Acta, 1995, 307, 109 CrossRef CAS.
- J. Zheng, M. Ohata and N. Furuta, J. Anal. At. Spectrom., 2002, 17, 730 RSC.
- J. M. Marchante-Gayón, I. Feldmann, C. Thomas and N. Jakubowski, J. Anal. At. Spectrom., 2001, 16, 457 RSC.
- M. A. Quijano, A. M. Gutiérrez, M. C. Pérez-Conde and C. Camara, Talanta, 1999, 50, 165 CrossRef CAS.
- B. Gammelgaard, L. Bendahl, U. Sidenius and O. Jøns, J. Anal. At. Spectrom., 2002, 17, 570 RSC.
- B. Gammelgaard, O. Jøns and L. Bendahl, J. Anal. At. Spectrom., 2000, 16, 339 Search PubMed.
- B. Gammelgaard, K. D. Jessen, F. H. Kristensen and O. Jøns, Anal. Chim. Acta, 2000, 404, 47 CrossRef CAS.
- J. M. G. Lafuente, M. L. F. Sánchez and A. Sanz-Medel, J. Anal. At. Spectrom., 1996, 11, 1163 RSC.
- B. Gammelgaard and O. Jøns, J. Anal. At. Spectrom., 2000, 15, 945 RSC.
- A. Chatterjee, Y. Shibata and M. Morita, J. Anal. At. Spectrom., 2000, 15, 913 RSC.
- T. H. Cao, R. A. Cooney, M. M. Woznichak, S. W. May and R. F. Browner, Anal. Chem., 2001, 73, 2898 CrossRef CAS.
- B. Gammelgaard, K. G. Madsen, J. Bjerrum, L. Bendahl, O. Jøns, J. Olsen and U. Sidenius, J. Anal. At. Spectrom., 2003, 18, 65 RSC.
- S. G. Patching and P. H. E. Gardiner, J. Trace Elem. Med. Biol., 1999, 13, 193 Search PubMed.
- J. M. G. Lafuente, M. Dlaska, M. L. F. Sánchez and A. Sanz-Medel, J. Anal. At. Spectrom., 1998, 13, 423 RSC.
- J. M. G. Lafuente, J. M. Marchante-Gayón, M. L. F. Sánchez and A. Sanz-Medel, Talanta, 1999, 50, 207 CrossRef CAS.
- L. Bendahl, B. Gammelgaard, O. Jøns, O. Farver and S. H. Hansen, J. Anal. At. Spectrom., 2001, 16, 38 RSC.
- S. J. Foster and H. E. Ganther, Anal. Biochem., 1984, 137, 205 CrossRef CAS.
- T. W. M. Fan, A. N. Lane, D. Martens and R. M. Higashi, Analyst, 1998, 123, 875 RSC.
- J. Zheng, Y. Shibata and A. Tanaka, Anal. Bioanal. Chem., 2002, 374, 348 CrossRef CAS.
- T. Lindemann, A. Prange, W. Dannecker and B. Neidhart, Fresenius’ J. Anal. Chem., 2000, 368, 214 CrossRef CAS.
- S. H. Bügel, E. H. Larsen, J. J. Sloth, K. Flytlie, K. Overvad, L. C. Steenberg and S. Moesgaard, unpublished results, 2003.
| This journal is © The Royal Society of Chemistry 2004 |