Separation and detection of Se-compounds by ion pairing liquid chromatography-microwave assisted hydride generation-atomic fluorescence spectrometry
Received
27th June 2003
, Accepted 9th October 2003
First published on 18th November 2003
Abstract
Liquid chromatography coupled to a hydride generation atomic fluorescence spectrometer has been applied for the speciation of Se in extracts of Saccharomyces cerevisiae. In order to develop a method which allows the separation of the compounds and detection of the element, seven Se standards were used: Se-methionine (Se-Met), Se-cystine (Se-(Cys)2), Se-cystamine (Se-Cya), Se-methylselenocysteine (Se-MeSeCys), Se-ethionine (Se-Et), selenate (SeVI), selenite (SeIV). Optimal chromatographic results were obtained with reversed-phase chromatography on an XTerra C18 column using a positively charged ion-pairing agent. It was observed that for these standards precise control of the pH was of utmost importance. Attention was devoted to the compatibility of the mobile phase with hydride generation. Efficient formation of the hydrides was obtained by optimisation of different parameters. The redox mixture which allowed optimum conversion of all different species was HBr–KBrO3. To assist in the conversion of the compounds, on-line microwave digestion was applied. The detection limits obtained for the standards were: 0.8 µg Se l−1 for selenite(IV); 1.3 µg Se l−1 for selenate(VI); 1.2 µg Se l−1 for Se-methionine; 1.2 µg Se l−1 for Se-cystine; 1.3 µg Se l−1 for Se-cystamine; and 1.1 µg Se l−1 for Se-methylselenocysteine, respectively. Se-compounds in Saccharomyces cerevisiae were extracted by hot water (50 °C) or proteolytic digestion with protease XIV (37 °C). The method developed for separation and elemental detection was applied to these extracts in order to distinguish between the different species extracted from the yeast matrix. Total Se concentration in the extracts was measured with pneumatic nebulization-inductively coupled plasma-mass spectrometry (PN-ICP-MS). Species transformation was investigated by analysing extracts preserved at 2 different temperatures (−20 °C and 4 °C). Only those extracts kept at −20 °C proved to be unchanged.
Introduction
It is known that selenium is an essential element for higher organisms. The dose at which selenium has to be taken in is very critical, since Se is essential only in a very narrow concentration range. Below this range, deficiency occurs, above it Se becomes toxic.1 Not only is the total amount of Se of importance to meet daily requirements. The form, or the chemical species, under which the element is supplemented determines its beneficial effect. It should be noted that the absorption of the element depends strongly on the chemical species in which it is consumed. Selenium compounds that are insoluble, or difficult to digest, are simply excreted by the body.2 Earlier research showed that plants take up Se as the inorganic form selenate(VI). When Se is incorporated in growing plants, it may replace sulfur in the amino acids.3 Dietary studies have shown that Se in amino acids is absorbed more readily by the body than an inorganic form of the element.4 Speciation of Se in food is therefore a very relevant issue. It is of the utmost importance to identify the active Se compounds because of their role in the human body. Their biochemical pathway throughout the body indicates that they possibly play a role in the battle against free radicals, and by doing so are active in the prevention of cancer. Because of this recognition, Se supplements have become very popular. The need for efficient control is growing as more and more suppliers commercialise Se-supplements.5 Yeast is often used as such a nutraceutical. Therefore there is an urgent need for a yeast reference material certified for its bioavailable Se-species.6,7 Because of the complexity of a yeast matrix, high resolution techniques become necessary to permit separation of, and distinction between, different compounds. Some researchers used CE-ICP-MS to separate and detect inorganic Se forms and the selenoamino acids. Detection limits obtained with this procedure for inorganic species and the amino acids were 10–20 and 30–50 µg l−1, respectively. Shifts in migration times were observed as more compounds were loaded on to the capillary.8 GC-ICP-MS has been used for the study of selenoamino acids and for chiral separations in, among other things, yeast.9,10 Others have used LC-ICP-MS to deal effectively with the specific needs of speciation analyses. The application of reversed-phase chromatography on a C18 or a C8 column with a mobile phase consisting of methanol in combination with an ion pairing agent (trifluoroacetic acid, heptafluorobutyric acid) has been reported.11,12 Attempts have been made to use 3-dimensional chromatography with ICP-MS and electrospray mass spectrometry (ES-MS) to identify the species present in yeast.5 Detection limits obtained in this combination were in the low µg l−1 range. Other groups have used anion and cation exchange chromatography to separate the Se compounds.13–15 Difficulties have been reported in the separation of the inorganic species when using reversed-phase chromatography.16 Also the combination of LC with hydride generation-atomic fluorescence spectrometry has been investigated. For hydride generation, different methods have been applied. UV light, microwave treatment and/or different reagent mixtures17–20 were used in order to convert the species into SeIV, the species needed for hydride formation.
In this paper, another approach was followed. For the separation of the Se-species, reversed-phase chromatography on an XTerra C18 column with the positively charged ion pairing agent tetraethylammonium chloride was applied. This allowed separation of all 7 species, in approximately 20 min with good resolution. Detection of the element by HG-AFS was investigated. The formation of hydrides was preceded using a HBr–KBrO3 mixture assisted with on-line microwave treatment. This method gave equivalent LODs for all compounds. The optimised method was applied for the speciation of Se in Se-enriched yeast. Efforts were made to develop an extraction procedure which at the same time was efficient and kept the species intact. Extraction procedures using water are reported to give recoveries of Se between 10 and 40%.21 While extractions using proteolytic enzymes give recoveries of about 70%.11 In this work, different extraction procedures were compared whereby special attention was devoted to possible differences in compounds extracted from the yeast matrix. Furthermore, the method was applied to investigate species transformation in the extracts. The total Se concentration in yeast tablets was determined with instrumental neutron activation analyses (INAA). Total Se concentration in the extracts was determined with pneumatic nebulisation-inductively coupled plasma-mass spectrometry (PN-ICP-MS).
Experimental
Chemicals
All reagents used were of analytical grade. Ultrapure Milli-Q water was produced in the laboratory with a Millipore system (Bedford, MA, USA). Protease from Streptomyces griseus, Se-standards (selenite, selenate, Se-methionine, Se-cystine, Se-cystamine, Se-ethionine and Se-(methyl)selenocysteine were obtained from Sigma–Aldrich (Bornem, Belgium). NaBH4
(98%) and tetraethylammonium chloride was also from Sigma–Aldrich. NaOH was purchased from Carlo Erba (Milan, Italy). HBr (48%) was from Merck (Darmstadt, Germany). KBrO3 was obtained from UCB (Brussels, Belgium). The selenium-enriched Saccharomyces cerevisiae analysed in this paper was Selenoprecise, selenium tablets, 100 µg, Pharma Nord (Velje, Denmark). Nitric acid 14 M was purified by sub-boiling distillation in quartz equipment.
Instrumentation
The LC system, equipped with an autosampler, was a Waters Alliance 2690 (Waters Corporation, Milford, MA, USA). All separations were done on a Waters XTerra MS C18 column (250 mm × 4.6 mm, 5 µm particles). An XTerra guard column (2.1 cm × 2 mm, 5 µm particles) was used to capture too strongly retained compounds present in the biological matrix. Both were from Waters Corporation. The atomic fluorescence spectrometer used was a PS Analytical Millenium Excalibur system, from Anatech (Heverlee, Belgium). For the element detection a Se super lamp, Photron (Victoria, Australia), was used. The Microwave Digester 301 used for digestion of the samples was from Prolabo (Fontenay-sous-bois, France). Air segmentation was performed by using one of the pumps on the PS Analytical system. ICP-MS experiments were done on a PerkinElmer SCIEX Elan 5000, quadrupole-based instrument (Glendale, Ontario, Canada). Instrumental neutron activation analysis (INAA) of total Se concentrations in yeast supplements was performed by irradiation in the Thetis nuclear reactor. All samples were irradiated for 60 s at a neutron flux of 1012 n cm−2 s−1. The 79Se radioisotope with a half life of 3.92 min and gamma irradiation energy of 95.9 keV was measured with a Ge-Li detector (Model 7229 Canberra series, Meriden, CT, USA).
Materials
Vials used for sample handling were from Alltech (Lokeren, Belgium). Sodium borohydride (98%) was filtered through 0.45 µm pore filters from Millipore (Bedford, MA, USA). All biological samples were filtered through 0.22 µm filters from Millipore prior to injection. All extraction procedures were carried out in polypropylene tubes of 5 ml from Falcon, Becton Dickinson Labware (Meylan, France). PFA tubings (0.5, 0.8, 1.0 mm id) were obtained from Alltech (Lokeren, Belgium).
Standards
Standard solutions of selenite, selenate, Se-methionine, Se-cystine, Se-cystamine, Se-ethionine and Se-methylselenocysteine were prepared at a concentration of 100 µg Se l−1. These solutions were used both for optimisation of the separation and of the hydride formation and AFS detection.
Standard solutions for ICP-MS measurements contained 100 µg Se l−1. Ga (final concentration 50 µg l−1) was added to each standard solution to act as an internal standard. Standards were diluted in 0.14 M HNO3. A calibration curve was constructed by measuring Se-standard solutions with concentrations of 10, 50, 100 and 500 µg Se l−1 in order to calculate total Se concentration in the extracts.
Results and discussion
Separation of the 7 Se-standards
In view of the intended HG-AFS detection, we decided to avoid the use of organic solvents in the mobile phase. It is known that organic solvents suppress the formation of hydrides. However, because of the nature of the compounds to be separated, we opted for reversed phase chromatography. However, the more hydrophobic compounds (Se-methionine and Se-ethionine) were retained too strongly on a conventional C18 column when no organic solvents were used. Even with the application of negatively charged ion pairing agents (trifluoroacetic acid or heptafluorobutyric acid), some compounds only eluted after 50 min. Furthermore, we were not able to separate the seven compounds with sufficient resolution. Since the organic compounds to be separated have an amphoteric character, shifts in pH can be usefully exploited in these experiments. From the pKa values of the inorganic compounds it is clear that at pH 4.5 the inorganic compounds are negatively charged. Under these conditions a positively charged ion pairing agent could be applied for the separation. The organic compounds had to be separated according to their hydrophobicity. Promising results were obtained using an XTerra column, which is a slightly modified C18 column. The XTerra column differs from normal silica based C18 columns in that it is manufactured in another way. Hydrophobicity is already distributed throughout the surface prior to derivatisation with hydrocarbon ligands. This results in fewer free silanol groups. Since the quality of peak shapes for highly basic compounds is dependent upon the concentration and activity of residual silanols, the XTerra columns are guaranteed to deliver the sharpest and most symmetrical peaks. Because of the lower number of free silanol groups XTerra columns also enable reduction of the analysis time and the use of a wider pH range. In this research there was special interest in the reduction of analysis time. The use of this column in combination with a mobile phase containing no organic solvents, and only a low percentage of tetraethylammonium chloride in combination with a precise adjustment of the pH of the mobile phase, enabled the separation of the seven Se standard with fully resolved peaks within 20 min (Fig. 1). The LC conditions are summarised in Table 1.
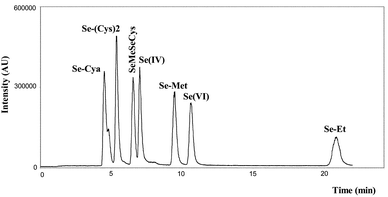 |
| | Fig. 1 Separation of 7 Se standards on Waters Xterra column. | |
Table 1 LC conditions for separation of 7 Se standards
| Column |
XTerra MS C18 |
| Column dimensions |
L 250 mm, id 4.6 mm, 5 µm |
| Guard column dimensions |
L 2.1 cm, id 2.0 mm, 5 µm |
| Mobile phase |
0.1% TEA+ Cl− in Milli-Q |
| pH |
4.0–4.5 |
| Injection volume |
20 µl |
| Sample loop |
100 µl |
| Flow rate |
0.8 ml min−1 |
Detection of Se by MW-HG-AFS
Since it was the intention to develop an on-line system which can be used for speciation analyses, it was of the utmost importance to preserve the chromatographic resolution. The time spent in the reaction coils by a compound eluting from the column prior to hydride formation is about 1 min. This results in major peak broadening. To cope with this problem, air segmentation was used. This was done by inserting air in the flow immediately after the column and ensured preservation of the chromatographic resolution.
Only SeIV is able to form hydrides. Hence, all compounds have to be converted into this oxidation state prior to detection. SeVI has to be reduced and the organic components have to be oxidised. The composition of the redox mixture used to efficiently convert all species to the suitable form is of great importance. A compromise was found in using HBr–KBrO3 assisted by on-line microwave oven heating. All optimised parameters for hydride generation are summarised in Table 2. Flow interruption, due to heating in the reaction coil, was observed after running the elution for approximately 8 min. By wrapping the coil around a spool in an ice bath after the microwave oven, this disturbance was overcome. The instrumental set-up is depicted in Fig. 2. Limits of detection were determined using the optimised conditions of Table 2. Se standard solutions of 100 µg l−1 Se were used (10 blank solutions of Milli-Q were measured as well). It was also important to verify the influence of the composition of the mobile phase on the signal of each compound. LODs were calculated both in the flow injection mode and in the separation mode (addition of XTerra column). The limits of detection were calculated according to the 3s criterion. The LODs in the flow injection mode were: 0.8 µg Se l−1 for selenite(IV); 1.3 µg Se l−1 for selenate(VI); 1.2 µg Se l−1 for Se-methionine; 1.2 µg Se l−1 for Se-cystine; 1.3 µg Se l−1 for Se-cystamine; and 1.1 µg Se l−1 for Se-methylselenocysteine, respectively. The LODs for the separation mode (use of Xterra C18 column and 0.1% TEACl as mobile phase) are presented in Table 3. Se-standards could be detected in the µg l−1 range. A remarkable observation was the response of Se-cystamine, as it appeared that this compound could only be detected in a narrow pH range around pH 4.5. The influence of the mobile phase was limited, as is clear from the LODs presented in Table 3. The detection limits obtained after separation were sufficient for speciation analysis in yeast. These conditions were applied for further research on Se-species in yeast extracts.
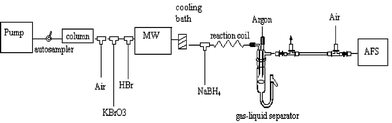 |
| | Fig. 2 Diagram of the applied instrumentation (LC-MO-HG-AFS). | |
Table 2 Conditions of MW-HG-AFS
| Air flow |
0.5 ml min−1 |
| Redox mixture |
HBr (48%)
+ 15 mM KBrO3 |
| Reducing agent |
1.4%
(w/w) NaBH4
+ 0.4%
(w/w) NaOH |
| HBr flow rate |
1.5 ml min−1 |
| NaBH4 flow rate |
1.5 ml min−1 |
| KBrO3 flow rate |
0.5 ml min−1 |
| Microwave power |
10 W continuously |
| Coil in MW |
L: 3 m; id: 0.8 mm |
| Coil in ice bath |
L: 2 m; id: 0.8 mm |
| Reaction coil |
L: 1.5 m; id 1.0 mm |
Table 3 Limits of detection (µg l−1) with MW HG-AFS detection after separation
| |
With XTerra column
|
| |
0.1% TEACl, pH 4.6 |
| Selenite |
4.2 |
| Selenate |
2.5 |
| Se-methionine |
8.3 |
| Se-cystine |
4.5 |
| Se-cystamine |
4.4 |
| Se-MeSeCysteine |
2.0 |
Different extraction procedures were compared in terms of their recoveries of total Se. Recovery was calculated versus the total concentration of Se as measured by INAA in the original product. According to these experiments, the total Se concentration of SePrecise yeast tablets was 328 ± 3 µg Se g−1 yeast. According to the supplier the concentration of Se per g of yeast was 332 µg. Each extraction was done using 0.2 g of yeast (ground with pestle and mortar to a homogenised powder) in 15 ml of Milli-Q water. Three different extraction procedures were compared. In procedure 1, the sample was put in a hot water bath at 50 °C for 24 h. The other 2 procedures were based on proteolytic digestion. Approximately 20 mg of protease XIV was added to 0.2 g of yeast in 15 ml Milli-Q water. In this second procedure, the sample was kept for 24 h at 37 °C in a hot water bath. In the third procedure, the sample was kept at 37 °C for 24 h in a shaking water bath at 150 rpm. After extraction the samples were centrifuged for 30 min at 6800×g and the supernatant was filtered through a 0.2 µm pore filter and frozen at −20 °C in order to avoid species transformation of the extracted substances. Prior to separation and Se determination the sample was defrosted prior to analysis.
Species transformation
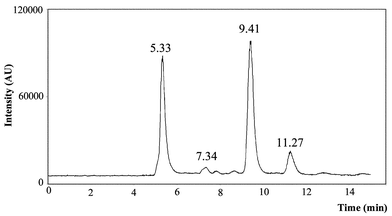
The stability of the species under certain conditions of preservation was questioned. The extract based on proteolytic digestion in a shaking water bath was first centrifuged. The supernatant was then filtered through a 0.2 µm pore filter. Part of the extract was preserved in the freezer at −20 °C for 3 weeks. Another part of the same extract was kept at 4 °C for 3 weeks. After that time the extracts were defrosted, filtered through a 0.2 µm pore filter and brought onto the column under the same separation and detection conditions as mentioned earlier. The chromatogram of the extract kept at −20 °C did not differ from the one shown in Fig. 3
(chromatogram 3). The chromatogram of the extract kept at 4 °C is shown in Fig. 4. From this chromatogram it is clear that severe species transformation occurs when the sample is kept at 4 °C. New peaks arise at retention times 5.33 min, 7.34 min and 11.27 min, which do not resemble any of the retention times of the applied Se-standards. Therefore for further experiments, samples will be preserved at −20 °C and only be defrosted prior to analysis. In order to check whether this transformation was due to the matrix of the yeast, Se-Met was treated in exactly the same way as the yeast sample. Therefore protease XIV was added to a Se-Met standard solution. This sample, together with a sample containing SePrecise and protease XIV, was shaken in a water bath for 24 h at 37 °C. Both samples were centrifuged, filtered and stored at 4 °C for 8 days. Separation and detection of the species showed that less than 1% of the Se-Met standard solution was transformed into another compound, whereas more than 11% of the Se-Met in SePrecise was converted. Therefore, we could conclude that the transformation is mainly caused by the yeast matrix, and not by the further action of protease. To trace the conversion of Se-Met by the protease action, Se-Met was added to SePrecise prior to enzymatic digestion. The first sample contained 80 mg of SePrecise, 3 ml of Milli-Q water and protease. The second sample contained 80 mg of SePrecise, 100 µl of a 100 mg l−1 Se-Met solution, 2.9 ml of Milli-Q water and protease. These samples were shaken during 24 h at 37 °C, centrifuged, filtered and stored at −20 °C. The compounds extracted were separated and detected. The result is shown in Fig. 5. The black curve represents the SePrecise sample: only one compound is detected here. The grey curve represents the SePrecise sample spiked with Se-Met. From this curve it is clear that Se-Met is not transformed by enzymatic hydrolysis, and that the retention times of the SePrecise compound and Se-Met are equal.
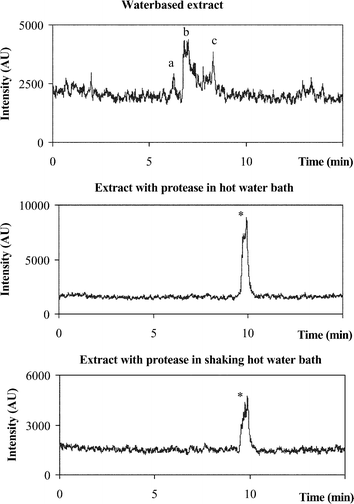 |
| | Fig. 3 Chromatograms of the 3 different extraction procedures a,b,c: species extracted by water *: compound extracted by protease XIV tR
= Se-Met. | |
 |
| | Fig. 4 Chromatogram of the proteolytic digest preserved in a refrigerator for 3 weeks. | |
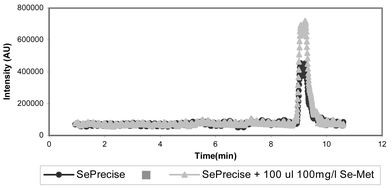 |
| | Fig. 5 Separation of the species extracted by proteolytic digestion, with addition of Se-methionine to demonstrate the stability of this species for protease XIV treatment. | |
Total Se concentration in the extracts
To measure the total amount of Se extracted by the three above mentioned extraction procedures pneumatic nebulisation-inductively coupled plasma-mass spectrometry (PN-ICP-MS) was used. Instrument parameters were as summarised in Table 4.
Table 4 PN-ICP-MS settings for determination of selenium
|
ICP-MS instrument
|
PerkinElmer Sciex Elan 5000
|
| Sample introduction flow |
0.75 ml min−1 |
| Rf Power |
1000 W |
| Ar gas flow rates |
|
| Plasma |
15 l min−1 |
| Auxiliary |
0.8 l min−1 |
| Nebulizer |
0.987 l min−1 |
| Data acquisition |
| Scanning mode |
Peak hopping |
| Points per spectral peak |
1 |
| Dwell time per acquisition point |
100 ms |
| Sweeps per reading |
100 |
| Readings per replicate |
1 |
| Replicates |
5 |
| Signals monitored |
69Ga+, 71Ga+, 82Se+ |
| Total measurement time |
3.5 min |
Since Se in the extracts is present in different forms, it is necessary to convert all compounds to the same extent. According to the detection limits obtained in Table 3, this was clearly not the case with HG-AFS in the flow injection mode. Variation of the signal between SeIV and the organic compounds was as high as 50%. For this reason it was impossible to measure total Se concentration in the extracts in which the identity and ratio of the species are not known. Therefore PN-ICP-MS was applied since this technique converts all species to the same extent, no matter what is the nature of the compound. Measurement of the Se in the different Se standards with PN-ICP-MS showed that the signal of the organic compounds varied from the signal of the inorganic compounds to a maximum of 2.5%. The detection limits were calculated according to the 3 s criterion. LODs were 0.03 µg Se l−1 for SeIV, Se-Met, Se-(Cys)2 and 0.04 µg Se l−1 for SeVI, Se-Cya and Se-MeSeCys, respectively. This variation is attributable to fluctuations of the ICP-MS signal. Some organic standards gave a higher signal while others gave a lower signal compared with the inorganic standards. For the determination of Se a 1 ml aliquot of the yeast extract was diluted in 0.14 M HNO3 to 25 ml. The recoveries of Se from yeast by the different extraction procedures were calculated versus the total concentrations measured with INAA. All total concentrations were measured 3-fold. The extraction efficiencies were: 19 ± 1% for water based extraction (1); 66 ± 1% for proteolytic digestion in a hot water bath (37 °C)
(2); and 86 ± 1% for proteolytic digestion in a shaking hot water bath (37 °C)
(3). It was clear that Se is released from yeast to a larger extent by proteolytic digestion in a shaking hot water bath. The question is, however, which species are extracted, and are these species the original compounds or transformations thereof. Therefore, identification of the species in the different extracts is necessary.
Separation of the different species extracted
Each extract was brought onto the XTerra column and eluted with 0.1% TEACl, pH 4.5, at a flow rate of 0.8 ml min−1. Se is detected with MW-HG-AFS according to the method described earlier. The chromatograms are shown in Fig. 3. From these chromatograms it is clear that different extraction procedures lead to different species. Although water extraction (chromatogram 1) results in the lowest recovery of Se, different species (at retention times 6.28 (a), 6.80 (b) and 8.30 (c)) can be distinguished in the chromatogram. These compounds are the unbound fraction of the yeast sample. The retention times of these species do not resemble any of the retention times of the used standards. Since in the proteolytic digests (chromatograms 2 and 3) only one Se-compound (*) was detected, the water extracts remain interesting. Their nature needs to be elucidated. The compound extracted by proteolytic digestion is the protein bound fraction of the yeast. The retention time of this compound resembled that of the Se standard Se-methionine. Standard addition of Se-methionine to the yeast extract showed that retention times of standard and yeast substance were indeed identical.
Conclusion
Ion pairing liquid chromatography coupled to hydride generation atomic fluorescence spectrometry with on-line microwave assistance is a suitable method for speciation analyses of complex biological matrices such as extracts of Saccharomyces cerevisiae. This method allowed us to distinguish the compounds extracted by different procedures. This combination is suitable for screening biological samples for the presence of Se-species and to screen the Se-compounds that are present in a matrix. This technique also admits to trace species transformations in a sample.
The extraction procedure based on enzymatic digestion gave the best Se recoveries. It was, however, necessary to determine the total Se concentration in the extracts by ICP-MS. Of course neither MW-HG-AFS, nor ICP-MS, permits species identification. This will be accomplished in later work by using electrospray tandem mass spectrometry.
Acknowledgements
E.D. is a grant holder of the Institute for the Promotion of Innovation by Science and Technology in Flanders (IWT). The authors express their gratitude to Dr. A. Rodriguez and Dr. G. Bordin of the IRMM, EC, Geel, Belgium for the loan of the Prolabo Microdigest 301. The authors also want to thank Louis Mees for carrying out the INAA experiments.
References
- T. Ferri, C. De Luca and L. Ticconi, Anal. Lett., 2001, 34(6), 975–988 CrossRef CAS.
- M. Stadlober, M. Sager and K. Irgolic, Food Chem., 2001, 73, 357–366 CrossRef CAS.
- C. C. Chéry, E. Dumont, R. Cornelis and L. Moens, Fresenius’ J. Anal. Chem., 2001, 371, 775–781 CrossRef CAS.
- A. Vonderheide, K. Wrobel, S. Kannamkumarath, C. B'Hymer, M. Montes-Bayon, C. Ponce de Leon and J. Caruso, J. Agric. Food Chem., 2002, 50, 5722–5728 CrossRef CAS.
- S. McSheehy, F. Pannier, J. Szpunar, M. Potin-Gautier and R. Lobinski, Analyst, 2002, 127, 223–229 RSC.
- R. Cornelis, H. Crews, O. Donard, L. Ebdon and Ph. Quevauviller, Fresenius' J. Anal. Chem., 2001, 370, 120–125 CrossRef CAS.
- H. Chassaigne, C. C. Chéry, G. Bordin and A. Rodriguez, J. Chromatogr. A, 2002, 976, 409–422 CrossRef CAS.
- S. Kannamkumarath, K. Wrobel, K. Wrobel, C. B'Hymer and J. Caruso, J. Chromatogr. A, 2002, 975, 245–266 CAS.
- C. Devos, K. Sandra and P. Sandra, J. Pharm. Biomed., 2002, 27, 507–514 Search PubMed.
- A. Sanz-Medel and E. Blanco-Gonzalez, Trends Anal. Chem., 2002, 21, 709–716 CrossRef CAS.
- M. Montes-Bayon, E. G. Yanes, C. Ponce de Leon, K. Jayasimhulu, A. Stalcup, J. Shann and J. A. Caruso, Anal. Chem., 2002, 74, 107–113 CrossRef CAS.
- J. Ruiz Encinar, R. Ruzik, W. Buchmann, J. Tortajada, R. Lobinski and J. Szpunar, Analyst, 2003, 128, 220–224 RSC.
- E. H. Larsen, M. Hansen, T. Fan and M. Vahl, J. Anal. At. Spectrom., 2001, 16, 1403–1408 RSC.
- M. Kotrebai, M. Birringer, J. Tyson, E. Block and P. Uden, Analyst, 2000, 125, 71–78 RSC.
- M. Vilano and R. Rubio, J. Anal. At. Spectrom., 2000, 15, 177–180 RSC.
- J. Zheng, M. Ohata and N. Furuta, J. Anal. At. Spectrom., 2002, 17, 730–735 RSC.
- I. Ipolyi, W. Corns, P. Stockwell and P. Fodor, J. Autom. Method Manag., 2001, 23, 167–172 Search PubMed.
- L. Pitts, A. Fisher, P. Worsfold and S. Hill, J. Anal. At. Spectrom., 1995, 10, 519–520 RSC.
- M. Johansson, G. Bordin and A. Rodriguez, Analyst, 2000, 125, 273–279 RSC.
- I. Ipolyi, Zs. Stefanka and P. Fodor, Anal. Chim. Acta, 2001, 435, 367–375 CrossRef CAS.
- M. Dernovics, Zs. Stefanka and P. Fodor, Anal. Bioanal. Chem., 2002, 372, 473–480 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2004 |
Click here to see how this site uses Cookies. View our privacy policy here.