Analysis of arsenic species accumulation by plants and the influence on their nitrogen uptake
Anne-Christine
Schmidt
a,
Werner
Reisser
a,
Jürgen
Mattusch
b,
Rainer
Wennrich
b and
Klaus
Jung
b
aUniversity of Leipzig, Institute of Botany, Johannisallee 22-24, 04103 Leipzig, Germany
bUFZ–Centre for Environmental Research Leipzig-Halle, Permoserstrasse 15, 04318 Leipzig, Germany
First published on 10th December 2003
Abstract
Terrestrial plants are able to accumulate arsenic to a substantial extent but survive the stress to differing degrees of vitality. The influence of arsenic on important energy and metabolic cycles does not yet have sufficient explanation. Parallel to the uptake and processing of arsenic species such as As(III) and As(V) by Silene vulgaris, the nitrogen uptake using a 15N tracer method was investigated. The results showed that the nitrogen uptake decreases with increasing arsenate concentrations applied to the plants. The reaction of the plants treated with arsenite changed from a depression at low arsenite concentrations to a strong increase with the largest quantity applied, exceeding the 15N-incorporation of the control plants. This behaviour underlines the divergent behaviour of the N-metabolism caused by both arsenic species. As(III) can be detoxified by complexation with peptides rich in SH-groups. As(V) acts as a phosphate analogue and interrupts diverse phosphorylation reactions.
Introduction
Arsenic contaminations of soils, sediments and waters occur from diverse anthropogenic and geogenic sources. The situation becomes particularly problematic in the regions of West Bengal and Bangladesh, where several millions of humans are exposed to arsenic contaminated drinking, ground and surface water.1–3 In Europe the problem of arsenic contamination particularly concerns, above al,l waste dumps of the mining industry as well as soils in military areas contaminated by ordnance containing arsenic compounds.4 In addition, some agriculture used regions are contaminated by the extensive use of arsenic compounds such as pesticides.5–8Terrestrial plants are able to accumulate arsenic to a substantial extent.9–11 So-called hyperaccumulators take up more than 1000 mg kg−1 dry weight of the pollutant.9 Two kinds of fern were found to be hyperaccumulators for arsenic: Pityrogramma calomelanos (ca. 8000 mg As kg−1) and Pteris vittata (ca. 6000 mg As kg−1). Since these plants bear such high arsenic quantities, they must have a strategy for detoxification.10
Very little is known about the processing of arsenic within the plant or tolerance mechanisms in view of high arsenic contents in the soil. Furthermore, no concrete data exists concerning the effect of arsenic on the metabolism of plants. Only sporadic evidence exists of growth impairments as a result of arsenic contamination.12,13 From more recent results it is assumed that a synthesis of peptides rich in thiol groups is induced with exposure to inorganic arsenic species.14–17
Since the element arsenic is subjected to various metabolisation reactions in biological systems, which can give rise to innocuous compounds, the total arsenic content alone does not offer sufficient information on the degree of toxicity. Therefore, the differentiation between different arsenic compounds (species) forms the basis for an exact evaluation of the toxicological potential of environmental samples (plants, soils, waters).
Different arsenic species vary in their solubility, mobility, bioavailability and phytotoxicity. Hence, studies concerned with the uptake of different arsenic species, both inorganic and organic, by plants and their effects on plant growth and nutrition, are of essential importance for the understanding of the behavior of arsenic in the system soil–plant.18
A multiplicity of plant types selected on the basis of ecological aspects were used for test series about growth and arsenic accumulation behavior on soil substrates containing tailings material.19 This material has a high arsenic load (200–600 mg kg−1 dw) and came from a waste dump of the tin ore mining industry in the Erzgebirge mountain range, Germany. In the context of the test series mentioned above, one plant, the Common Campion (Silene vulgaris), showed a high arsenic accumulation and a simultaneous arsenic tolerance. This tolerance obviously results from its ability to reduce pentavalent arsenic, because (compared with other non-tolerant plants, for example grass and herbs) Silene vulgaris possesses strongly increased concentrations of trivalent arsenic. On the other hand, in animal experiments the trivalent form of inorganic arsenic has proved to be more toxic than the pentavalent form.20,21 Obviously, in plant organisms other effect mechanisms exist for ter- and pentavalent arsenic in comparison to animal organisms.
In the current project the uptake and processing of As(III) and As(V) by Silene vulgaris was investigated. For this purpose parallel exposure series with different concentrations of As(III) and As(V) were performed. The total arsenic contents of the plants were analyzed by hydride generation atomic absorption spectrometry (HG-AAS), and their arsenic species contents by ion chromatography coupled with inductively coupled plasma mass spectrometry (IC-ICP-MS). For the extraction of the arsenic species an optimized variant of accelerated solvent extraction (ASE) was used.22
The effect of the different levels of arsenic contamination on the plant metabolism was examined by means of the 15N-tracer technique.23–26 In plant cells, nitrogen plays an important role as a main component of amino acids, proteins and other biomolecules. Therefore, nitrogen metabolism, including the uptake and assimilation of nitrate, is one basis for an optimal biosynthesis of amino acids and proteins, which are involved in nearly all processes in plants.27 Because of the important role of nitrogen metabolism in plants, the assessment of the toxicity of arsenic with reference to this pathway is a basic step for elucidating the arsenic effect on plant organisms.
Experimental procedures
Plant cultivation, arsenic and tracer application
Plant cultivation and arsenic exposition took place in a phytochamber (Bioline Pflanzenwuchsschrank Typ VB 1514, Vötsch Industrietechnik GmbH, Balingen, Germany) under controlled light (60 W m−2, day duration 13 h), temperature (21 °C day, 15 °C night) and humidity (60% day, 75% night) conditions. The seeds were obtained from the Institut für Pflanzengenetik und Kulturpflanzenforschung Gatersleben, Germany. After sowing in pots with soil (800 g dw per pot) the seedlings were fed for 2 weeks with water. Then, an aqueous solution of the arsenic species was added at regular intervals over 4 weeks. Arsenite (As(III)) and arsenate (As(V)) were applied in three concentrations (15 mg L−1, 30 mg L−1, 60 mg L−1). For reproducibility, 5 pots containing 15 plants were used for each concentration. The soil used can be characterized by the following parameters: Substrat Typ 1 for plants with low or medium nutrient demand, mixture of moderately and strongly decomposed high moorland peat (H2-H5 and H6-H8), salt content <1.5 g l−1, pH (CaCl2) = 5.0 to 6.5, N 175 mg l−1, P2O5 190 mg l−1, K2O 240 mg l−1.In the irrigation water dissolved 15N-marked KNO3 (20 mg N in 20 ml KNO3 solution per 400 g dry soil, 10 at% K15NO3) was given as tracer 2 d before the harvest of the plants for the investigation of the N-uptake capacity.
Sample preparation for the determination of arsenic species and total arsenic concentrations in plants
After exposure to arsenic the plants were harvested and washed with tap water and, finally, with distilled water. The plants were divided into roots, stems, leaves and, if available, blooms. The different parts of plants were cut up homogeneously with a mortar and pestle under liquid nitrogen. Then they were weighed to 0.5–2 g in 11 ml-extraction cells for ASE (accelerated solvent extraction, Dionex-200, Sunnyvale, USA). Parallel to this the dry weight of the appropriate fresh samples was determined by drying process in a furnace at 90 °C.The ASE parameters selected after an optimization are used as follows.
Solvent: bidistilled water; pressure: 100 bar; temperature: 120 °C; static time: 10 min; purging: 60% of cell volume; number of cycles: 1; purge time: 180 s.
The received aqueous extract was filled up to 50 ml and then analyzed within 24 h (the stability of the samples is limited to this time) to determine its arsenic species contents and, after stabilization by acidifying with 32% HCl (500 µl acid on 4.5 ml sample), for its total arsenic content.
For the determination of the total arsenic contents without previous extraction the plant samples were subjected to a microwave digestion (Anton Paar Multiwave, PerkinElmer). After the drying process in the furnace at 90°C the plant samples were ground to a fine powder with an ultracentrifugal mill (ZM1, Retsch, Germany) and weighed in portions of approximately 500 mg into microwave-transparent digestion containers. Extraction residues of the accelerated solvent extraction (ASE, see above) were dried and weighed without further cutting up into the digestion containers.
For the determination of total arsenic concentrations 0.5 ml of 30% H2O2 (Merck) and 5 ml of 65% HNO3 (p.A., Merck) were added to each sample. The microwave digestion was carried out by a staged program (80 min, maximum pressure: 75 bar, maximum power 1000 W).
The digested samples were placed in 50 ml calibrated flasks and filled up to the calibration mark with H2O (MilliROPlus10, Millipore). The determination of total arsenic was carried out with HG-AAS.
Sample preparation for the determination of arsenic species and total arsenic concentrations in the water soluble fraction of the soils
Immediately after the harvest of the plants the soil in the pot was homogenized by agitation. Fresh samples were divided into 10 g portions and shaken in 50 ml of distilled water for 2 h. Analogously to the plant samples, the determination of the dry weight was realized via a drying process of 3 representative mixed samples at 105 °C.The extracts were analyzed regarding their arsenic species composition with IC-ICP-MS as well as their total arsenic concentrations with HG-AAS.
Determination of arsenic species with IC-ICP-MS
For the determination of arsenic species the method developed by Londesborough et al.28 was used. The separation of different arsenic species was performed by ion chromatography (separation column IonPac AS7 (Dionex) in combination with a guard column IonPac AG7 (Dionex); injection volume: 200 µl). A gradient program with 2 eluents (eluent A, 50 mM HNO3; eluent B, 0.4 mM HNO3) was used. The coupling was carried out with a quadrupole mass spectrometer (ELAN 5000, PerkinElmer) for element-specific detection at m/z =75 via a cross-flow nebulizer.Determination of total arsenic concentrations with HG-AAS
Since in the resulting samples inorganic arsenic compounds were present almost exclusively, it was possible to use HG-AAS for total arsenic determination, without having to consider the problems arising with organic compounds owing to incomplete hydride formation.29–31A combination of an FIAS 400 for hydride generation in the flow injection mode and an atomic absorption spectrometer (ZL 4100, PerkinElmer) with an electrothermal atomizer was used. The hydrides were enriched in a graphite furnace (permanently modified with Pd). The absorbance was measured at the wavelength 193.7 nm.
Arsenic hydride was produced by a pre-reduction of As(V) with potassium iodide and ascorbic acid and by nascent hydrogen. The nascent hydrogen was formed by acidifying NaBH4 with HCl.
Sample preparation for 15N-analysis
For sample preparation for the 15N tracer analysis the micro-Kjeldahl technique was used.32 0.1–0.5 g of freshly harvested leaves were digested with 2 ml of nitrogen-free sulfuric acid (H2SO4 conc.) by heating until the solutions were clear. The resulting (15NH4)2SO4 solutions were distilled using a modified micro-Kjeldahl-apparatus. The amount of nitrogen in the sample was determined by titration against a 0.02 N H2SO4 standard solution. Then, the samples were dried in a water bath, and the pure ammonium salts were collected.Analysis of the 15N isotope by emission spectrometry
The emission spectrometric measurement of 15N abundance (15N at%) of the nitrogen fraction of Silene vulgaris was performed with a 15N analyzer system (NOI-7, Fischer Analyseninstrumente, Leipzig, Germany). An amount of 10 µg of nitrogen as (15NH4)2SO4 dissolved in 30 µl of distilled water was used. After measuring the 15N abundance, the 15N excess abundance (15N at% exc.) was calculated by subtraction of the natural 15N abundance of 0.366 at%.Results and discussion
Arsenic species in soils used for plant cultivation
By using pure water as an extracting agent the existing relationship between the arsenic compounds in the soil is not changed, so that the arsenic extracted represents the element fraction actually existing in the soil solution. In the water extracts of the soils treated with inorganic arsenic (arsenite or arsenate), only these two species were found. Organic arsenic compounds did not occur. In an aerobic soil environment the thermodynamic equilibrium between tri- and pentavalent arsenic is greatly on the side of the more highly oxidized species. Thus, trivalent arsenic, which was added to the soil, is subject to a rapid oxidation, which was stopped within 3–5 d in previous experiments.33 During irrigation a continuous As(III) supply on all 2 or 3 days ensured an availability of As(III) to plants separately from a constantly rising As(V) pool. Up to the time of the plant harvest the applied As(III) was oxidized completely to As(V), as is shown by IC-ICP-MS analysis of the soil extracts. The oxidation of As(III) added to the soil during plant cultivation represented a problem in other studies34 as well, as at the end of the experiment the proportions of the arsenic species between As(III) and As(V) treatments did not differ in the soil.Dependence of arsenic accumulation in Silene vulgaris on the oxidation state of the applied arsenic
The behaviour of the arsenic accumulation of Silene vulgaris amended with the same concentrations, but different redox species, of inorganic arsenic are represented in Fig. 1 a–d.Obvious graduations of the plants treated with different As concentrations can be seen. With an increasing quantity of applied arsenic the uptake by plants rose. Compared with the control plants cultivated without arsenic, the tissue arsenic concentrations of all plant parts are increased. Plants treated with As(III) accumulated more arsenic than plants amended with As(V), especially in the case of stems and green leaves.
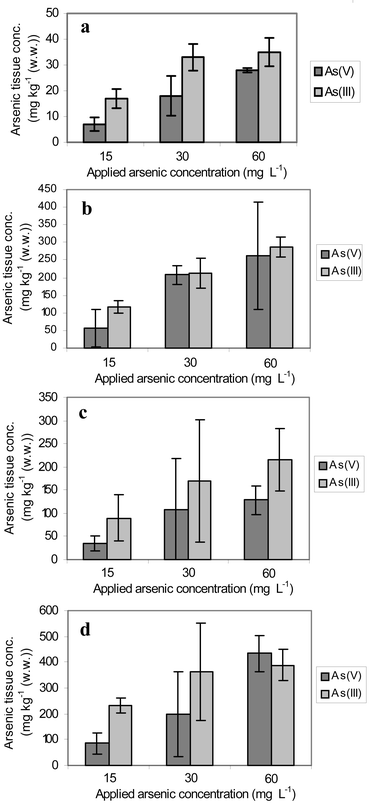 | ||
| Fig. 1 Dependence of arsenic concentrations accumulated in different plant parts on the applied quantity of arsenic and on its oxidation state. a, Green leaves: mean value and standard deviation from 5–12 parallel samples; arsenic concentration in leaves of control plants without arsenic application: 0.1 mg kg−1 w/w. b, Dead leaves: mean value and standard deviation from 3–5 parallel samples; arsenic concentration in dead leaves of control plants without arsenic application: 0.5 mg kg−1 w/w. c, Stems: mean value and standard deviation from 3–5 parallel samples; arsenic concentration in stems of control plants without arsenic application: 0.1 mg kg−1 w/w. d, Roots: mean value and standard deviation from 3–4 parallel samples; arsenic concentration in stems of control plants without arsenic application: 0.1 mg kg−1 w/w. | ||
The standard deviations show the biological range of the As accumulation. In stems a large fluctuation of the arsenic concentrations exists. The stems act as systems for substance transport. Therefore, it cannot be assumed that there is a uniform concentration status. A wide spreading of plant arsenic concentrations also occurs in natural ecosystems. In an arsenic contaminated area the arsenic concentrations in plants of the same location and the same kind varied very strongly from sample to sample.10 In the different organs of the plants the arsenic was stored to a greatly differing extent (Fig. 2). From the view of the arsenic distribution on the different organs it is possible to derive that a large part of the arsenic taken up by the roots also remains in it. Further large quantities are transferred into the stems. A transportation barrier to green leaves exists, which is destroyed in dead leaves. Hence, the As concentrations in dead leaves are 7–12 times higher than in intact leaves. Some individuals developed blooms. The arsenic content in these blooms was very small despite the very high arsenic concentrations in the other plant parts (6–7 mg kg−1 (w/w)). A transport of arsenic from stem into blooms does not usually take place. Generally, a uniform pattern appears concerning the arsenic distribution on the different organs of the plant: the highest arsenic concentrations are located in the roots and the lowest in fruits and seeds, while the concentrations in the vegetative parts above ground lie in between.35–38
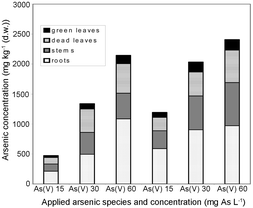 | ||
| Fig. 2 Distribution of arsenic on different organs of Silene vulgaris after application of As(III) and As(V) at three concentrations. Fig. 3. | ||
Metabolisation of ingested arsenic in the plant organism dependent on the applied arsenic oxidation state and location inside the plant
The analysis of the arsenic species by IC-ICP-MS showed that a metabolisation of the inorganic arsenic taken up by the terrestrial plants did not normally take place. In the chromatograms some additional small peaks occurred, which are shown in the amplified view of Fig. 3. However, the appropriate As concentrations are very low, only 0.39 ± 0.08% for green leaves, 0.2% for brown leaves, 0.02% for stems and 0.02% for roots (data given for As(III)-treated plants, 60 mg l−1) in relation to the two dominating species As(III) and As(V). The retention time of a peak (between 252 and 302 s) corresponds with the retention time of the DMA standard. It was not possible to assign all the other peaks to the available standard compounds (MMA, AsB, AsC; TMAO, TMAs+). Here, the widespread dominance of inorganic As-species in terrestrial plants is asserted.39,40 Higher terrestrial plants do not use the methylation pathway of inorganic arsenic postulated for marine animals and algae, which leads from simple methylated species up to arsenosugars, arsenocholine and arsenobetaine.41–43 Recently, arsenosugars were also found in shells,44 in earthworms45 and in fresh water plants.46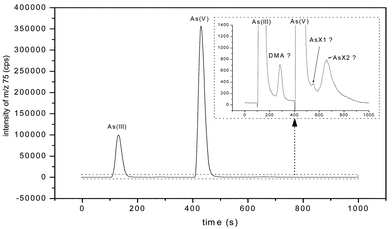 | ||
| Fig. 3 Ion chromatogram (IC-ICP-MS) and amplified view of a water extract of dead leaves. Plants treated with As(III), 30 mg l−1. AsX1, AsX2: unidentified peaks. | ||
Current investigations showed that the methylated trivalent species appearing as intermediate products of the methylation pathway are at least as toxic or somewhat more toxic than the inorganic species.47,48 These new findings concerning the toxic intermediate metabolites (MMAIII and DMAIII) raise doubts as to whether the biomethylation of arsenic is really a detoxification process. Some mammals do not methylate inorganic arsenic either, implying that the methylation cannot be the only way of detoxification.49–51
The redox status of the dominating inorganic arsenic varied between different organs (Fig. 4).
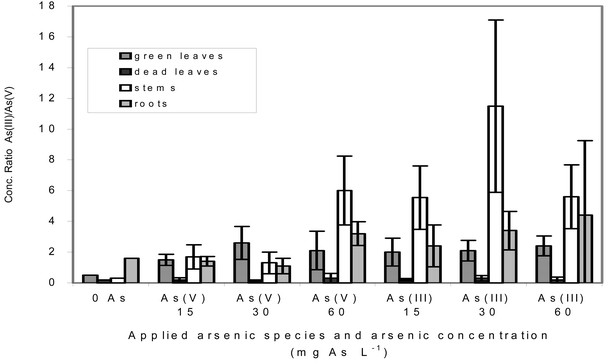 | ||
| Fig. 4 Dependence of the As(III) ∶ As(V) concentration ratios in different plant organs on the applied concentration and redox form of arsenic. Mean value ± standard deviation from n parallel samples: n = 5–12 for green leaves, n = 4–5 for dead leaves and for stems, n = 3–5 for roots. | ||
Independently of the applied redox form and of the applied quantity of arsenic, approximately double the quantity of As(III) compared with As(V) was found in green leaves. This means a shift of the As(III) ∶ As(V) ratio in favor of the reduced form compared with control plants. In stems also the As(III) ∶ As(V) ratio increased during arsenic exposure compared with unexposed plants. This rise was stronger in plants treated with As(III) than in plants treated with As(V). Altogether, the highest As(III) concentrations were achieved in stems. Nothing is known about the mechanisms of the reduction of arsenate in plants. In yeast cells a specific enzyme, arsenate reductase, catalyzes the reduction from As(V) to As(III). The regeneration of the arsenate reductase oxidized thereby takes place via glutathion and glutaredoxin.52 In bacteria an enzyme known as ArsC catalyzes the reduction from arsenate to arsenite. The enzymatic reaction needs reduced glutathion and glutaredoxin here also.53
The analysis of the arsenic species in dead leaves, separated from the green leaves, affirmed the hypothesis set up on the basis of previous experiments,19 that in dead plant parts the pentavalent form of inorganic arsenic dominated. The As(III) ∶ As(V) concentration ratio in dead leaves is very small (nearly zero). The very high total arsenic concentrations are caused by arsenate only.
Already in roots high As(III) quantities were found, suggesting a strong reduction ability of root cells in view of the dominating pentavalent redox form in the soil.
Since arsenate acts as a phosphate analogue during the transport through the plasma membrane of root cells,54 is it highly probable that an arsenate uptake occurs. On the other hand, an uptake of trivalent arsenic is not to be excluded, especially in the case of the plants amended with As(III), since a new quantity of As(III) is added to the soil with each application step. Its complete oxidation occurs within 3–5 d, as was mentioned above. In addition, microorganisms ocurring in the rhizosphere and/or the excretion of substances by the roots could lead to a reduction of arsenate in the direct root vicinity. Tluštos et al.34 also ascertained a dominance of trivalent arsenic in the roots from radish plants both after As(III) and As(V) treatment. Despite an exclusive supply of As(V), the As(III) portion is at least as large as the As(V) portion in living plant tissues. Also, in roots and spears of pine seedlings after arsenate exposure arsenite was found to be the main species.55 The plant may reduce As(V) because the trivalent arsenic species can be bound by peptides with SH-groups and thereby detoxified. Langdon et al.56 examined intact tissues of earthworms from arsenic-contaminated soils with X-ray absorption spectroscopy and found As(III)
in coordination with 3 groups of sulfur. The coordinating molecules are probably proteins rich in SH-groups such as metallothioneines. Such S–As(III) coordinations are not stable under non-physiological conditions and break during methanolic extractions. This could explain the high concentrations of toxic arsenite found in tissue extracts. There has been no clarification as to whether extractions with pure water also destroys such As(III)–sulfur coordinations. In the plant extracts arsenic available as As(III) could be dissociated from As(III)–thiol complexes.
Extractability of the accumulated arsenic—evaluation of the extraction efficiency
The extraction efficiency (%) was calculated from the quotient of the concentration extracted by ASE multiplied by 100 and the sum of the ASE concentration and the concentration in the residue of extraction. The extraction efficiencies are 87 ± 12% (As(V) amended) and 94 ± 3.8% (As(III) amended)(Table 1).The extractability of arsenic is independent of both the applied arsenic quantity and the quantity accumulated in the different tissues. The small quantities of non-extracted arsenic can be bound strongly to lipids or cell wall components such as cellulose, calcium and magnesium pectates and lignin.46 Nothing is known about such linkages of arsenic species. Furthermore, lipid soluble arsenic species are not extractable with the extracting agent water. Any occurrence of lipid soluble arsenic species in higher plants is still unknown. In brown algae lipid soluble arsenic amounted to only 3–4% of the total arsenic.57
Influence of the applied arsenic species on the capacity of nitrogen metabolism
Using K15NO3 as a nitrogen source for the plants, the nitrogen incorporation into the leaves was determined by means of the 15N tracer technique for the investigation of the activity of the nitrogen metabolism. If a pollutant disturbs the function of proteins (enzyme functions, structure functions, regulator functions), the plant tries to compensate for the loss of the function by increasing the protein resynthesis. In addition, a synthesis of special stress enzymes can occur. The protein turnover rises. If the toxicity of the pollutant is too great, the defence mechanisms are hindered and a general degeneration of the metabolism occurs. The protein synthesis and thus the metabolic activity decrease.The 15N-incorporation as a function of the applied redox form of arsenic and its amount is represented in Fig. 5. The application of As(III) causes a different reaction of the nitrogen metabolism from the application of As(V). With the latter a linear decrease (y = −0.32x + 1.21, R2 = 0.931) of the nitrogen uptake with a rising quantity of the applied arsenic is noted. In contrast, the As(III) application also led first to a depression of the 15N uptake, but caused a strong increase with the largest quantity applied, exceeding the 15N incorporation of the control plants. As an explanation for such an increase in the 15N abundance a stress-induced protein biosynthesis is possible. However, it remains unclear why this protein synthesis begins only after the greatest arsenic application. A possible explanation would be a change in the physiological processing of the arsenic taken up, which occurs with an increasing concentration in the soil environment.
In summary, As(III) has a less negative effect on the plant organism than As(V), because the plants treated with As(V) were strongly influenced so that they were not able to increase their protein synthesis for protection against the As damage at each of the three given concentrations. A stronger toxic effect of arsenate in comparison to arsenite for plants was also postulated by Koch et al.58 and by Cullen et al.59
According to the 15N incorporation all As applications differ significantly (**) to highly significantly (***) from 0 mg L−1 As controls (Table 2). Comparing the 15N incorporations of the same applied arsenic concentrations of the As(III) and As(V) series, significant (**) to highly significant (***) differences exist everywhere. This behaviour underlines the diverging behavior of the N-metabolism caused by both arsenic species. As(III) can be detoxified by complexation with peptides rich in SH-groups. As(V) acts as a phosphate analogue and interrupts diverse phosphorylation reactions, above all ATP production. Furthermore, it can pass membranes using phosphate transporters. Therefore, it can arrive in organelles, in particular in mitochondria, where it blocks components of the respiratory chain after reduction to As(III).
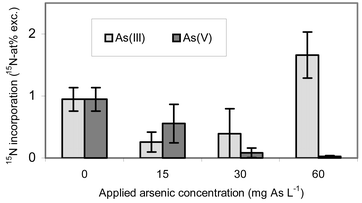 | ||
| Fig. 5 Uptake of the nitrogen isotope 15N into the leaves of Silene vulgaris after exposure to As(III) and As(V) at different concentrations. Mean value and standard deviation from n = 13 parallel samples. For the As(V) series the mathematical equation as well as the coefficient of correlation is specified. | ||
| Plant organ, number of parallels | As(V), 15 mg As L−1 | As(V), 30 mg As L−1 | As(V), 60 mg As L−1 | As(III), 15 mg As L−1 | As(III), 30 mg As L−1 | As(III), 60 mg As L−1 |
|---|---|---|---|---|---|---|
| Green leaves, n = 2–6 | 73 ± 1 | 86 ± 5 | 89 ± 7 | 92 ± 2 | 85 ± 8 | 89 ± 1 |
| Dead leaves, n = 1–3 | 91 | 95 | 94 ± 3 | 94 ± 5 | 96 ± 2 | 95 ± 2 |
| Stems, n = 1–3 | 88 | — | 95 ± 5 | 97 ± 3 | 95 ± 1 | 97 |
| Roots, n = 1–3 | 87 | — | 98 ± 2 | 97 ± 2 | 96 ± 1 | — |
| (a) | ||||||
|---|---|---|---|---|---|---|
| As(V), 15 mg As L−1 | As(V), 30 mg As L−1 | As(V), 60 mg As L−1 | As(III), 15 mg As L−1 | As(III), 30 mg As L−1 | As(III), 60 mg As L−1 | |
| 0 As (controls) | p < 0.01** | p < 0.001*** | p < 0.001*** | p < 0.001*** | p < 0.001*** | p < 0.001*** |
| (b) | |
|---|---|
| As concentration | Significance between As(III) and As(V) application |
| 15 mg As L−1 | p < 0.01** |
| 30 mg As L−1 | p < 0.01** |
| 60 mg As L−1 | p < 0.001*** |
References
- A. Ahmad, D. Bandaranayake, A. W. Khan, A. Hadi, G. Uddin and A. Halim, Int. J. Environ. Health Res., 1997, 7, 271–276 CrossRef.
- T. R. Chowdhury, G. K. Basu, B. K. Mandal, B. K. Biswas, G. Samanta, U. K. Chowdhury, C. R. Chanda, D. Lodh, S. L. Roy, K. C. Saha, S. Roy, Q. Quamruzzaman and D. Charaborti, Nature, 1999, 401, 545–546 CAS.
- R. Nickson, J. McArthur, W. Burgess, K. M. Ahmed, P. Ravenscroft and M. Rahman, Nature, 1998, 395, 338 CrossRef CAS.
- K. A. Feller, Arseneliminierung aus kontaminiertem Erdreich durch Bioakkumulation. Waste Management of Energetic Materials and Polymers, 23rd Annual Conference, Karlsruhe, Germany, 1992 Search PubMed.
- L. Bernstam and J. Nriagu, J. Toxicol. Environ. Health B, 2000, 3, 293–322 Search PubMed.
- L. R. Johnson and A.E. Hitbold, Soil Sci. Soc. Am. Proc., 1969, 33, 279–282 Search PubMed.
- I. Merwin, P. T. Pruyne, J. G. Ebel, K. L. Manzell and D. J. Lisk, Chemosphere, 1994, 29, 1361–1367 CrossRef CAS.
- P. Mitchell and D. Barr, Environ. Geochem. Health, 1995, 17, 57–82 CAS.
- R. R. Brooks, J. Lee, R. D. Reeves and T. Jaffre, J. Geochem. Explor., 1977, 7, 49–57 CrossRef CAS.
- P. Visoottiviseth, K. Francesconi and W. Sridokchan, Environ. Pollut., 2002, 118, 453–461 CrossRef CAS.
- R. Wagemann, N. B. Snow, D. M. Rosenberg and A. Luts, Arch. Environ. Contamin. Toxicol, 1979, 7, 169–191.
- E. A. Woolson, ACS Symp. Ser., 1975, 7, 176.
- A. A. Carbonell, M. A. Aarabi, R. D. DeLaune, R. P. Gambrell and W. H. Patrick Jr., Sci. Tot. Environ., 1998, 217, 189–199 Search PubMed.
- J. Hartley-Whitaker, G. Ainsworth, R. Vooijs, W. M. Ten Bookum, H. Schat and A. A. Meharg, Plant Physiol., 2001, 126, 299–306 CrossRef CAS.
- T. Maitani, H. Kubota, K. Sato and T. Yamada, Plant Physiol., 1996, 110, 1145–1150 CAS.
- M. E. V. Schmöger, M. Oven and E. Grill, Plant Physiol., 2000, 122, 793–801 CrossRef CAS.
- F. E. Sneller, L. M. Van Heerwaarden, F. J. L. Kraaijeveld-Smith, W. M. Ten Bookum, P. L. M. Koevoets, H. Schat and J. A. C. Verkleij, New Phytol., 1999, 144, 223–232 Search PubMed.
- A. A. Carbonell-Barrachina, F. Burló, D. Valero, E. López, D. Martínez-Romero and F. Martínez-Sanchez, J. Agric. Food Chem., 1999, 47, 2288–2294 CrossRef CAS.
- A. C. Schmidt, Diploma Thesis, University of Leipzig, Germany, 1999.
- B. Amran, F. Lagarde, M. J. F. Leroy, A. Lamotte, C. Demesmay, M. Ollé, M. Albert, G. Raurer and J. F. López-Sánchez in Quality Assurances for Environmental Analysis, eds. P. Quevauviller, E. A. Maier, B. Griepink, Elsevier Science BV., 1995, pp. 285–304 Search PubMed.
- H. Yamauchi and B. A. H. Fowler in J. O. Nriagu, ed., Arsenic in the environment, Part II: Human Health and Ecosystem Effects, John Wiley & Sons, New York, 1994, pp. 35–53 Search PubMed.
- A. C. Schmidt, W. Reisser, J. Mattusch, P. Popp and R. Wennrich, J. Chromatogr. A, 2000, 889, 83–91 CrossRef CAS.
- IAEA, Nitrogen-15 in Soil–Plant Studies, Panel Proceedings Series, International Atomic Energy Agency, Wien, 1971 Search PubMed.
- K. Jung, W. Rolle, D. Schlee, H. Tintemann, T. Gnauk and G. Schüürmann, New Phytol., 1994, 128, 505–508 Search PubMed.
- K. Jung, K. Kaletta, H. Segner and G. Schüürmann, ESPR-Environ. Sci. Pollut. Res., 1999, 6, 72–76 Search PubMed.
- K. Jung and P. Junghans, Biol. Zbl., 1981, 100, 217–226 Search PubMed.
- B. G. Forde and D. T. Clarkson, Adv. Bot. Res., 1999, 30, 1–90 Search PubMed.
- S. Londesborough, J. Mattusch and R. Wennrich, Fresenius’ J. Anal. Chem., 1999, 363, 577–581 CrossRef CAS.
- K. A. Francesconi and J. S. Edmonds, Adv. Inorg. Chem., 1997, 44, 147–189 CAS.
- S. Ringmann, K. Boch, W. Marquardt, M. Schuster, G. Schlemmer and P. Kainrath, Anal. Chim. Acta, 2002, 452, 207–215 CrossRef CAS.
- B. Welz and M. Melcher, Anal. Chem., 1985, 57, 427–431 CrossRef CAS.
- H. Faust, FAO/IAEA Interregional Training Course on the Use of 15N in Soil Science and Plant Nutrition, ZFI-Mitteilungen, Berichte des Zentralinstituts für Isotopen- und Strahlenforschung Leipzig, 1981, 38, 176.
- A. C. Schmidt, W. Reißer and J. Mattusch, Untersuchungen zur Schadstoffmobilisierung und -festlegung in Tailingsmaterialien durch Algen, Moose und Makrophyten, UFZ-Bericht 25/97, UFZ Leipzig-Halle GmbH, Leipzig, 2001 Search PubMed.
- P. Tlustoš, W. Goessler, J. Szákova and J. Balík, Appl. Organometal. Chem., 2002, 16, 216–220 CrossRef CAS.
- M. J. Abedin, M. S. Cresser, A. A. Meharg, J. Feldmann and J. Cotter-Howells, Environ. Sci. Technol., 2002, 36, 962–968 CrossRef CAS.
- A. A. Carbonell-Barrachina, F. Burló, D. Valero, E. López, D. Martínez-Romero and F. Martínez-Sanchez, J. Agric. Food Chem., 1999, 47, 2288–2294 CrossRef CAS.
- E. I. Hozhina, A. A. Khramov, P. A. Gerasimov and A. A. Kumarkov, J.Geochem. Explor., 2001, 74, 153–162 CrossRef CAS.
- N. W. Lepp, Effect of Heavy Metal Pollution on Plants, Effects of Trace Metal on Plant Function, Applied Science Publishers, London, 1981, vol. 1 Search PubMed.
- H. Helgesen and E. H. Larsen, Analyst, 1998, 123, 791–796 RSC.
- R. H. Pyles and E. A. Woolson, J. Agric. Food Chem., 1982, 30, 866–870 CrossRef CAS.
- H. V. Aposhian, R. A. Zakharyan, Y. Wu, S. M. Healy and M. M. Aposhian in Arsenic Exposure and Health Effects II, eds. C. O. Abernathy, R. L. Calderon and W. R. Chappell, Chapman & Hall, New York. 1997, pp. 296–321 Search PubMed.
- H. V. Aposhian, R. A. Zakharyan, E. K. Wildfang, S. M. Healy, J. Gailer, T. R. Radabaugh, G. M. Bogdan, L. A. Powell and M. M. Aposhian in Arsenic Exposure and Health Effects III, ed. W. R. Chappell, C. O. Abernathy and R. L. Calderon, Elsevier Science, New York, 1999, pp 289–297 Search PubMed.
- W. R. Cullen and K. J. Reimer, Chem. Rev., 1989, 89, 713–764 CrossRef.
- E. H. Larsen, Fresenius’ J. Anal. Chem., 1993, 352, 582–588 CrossRef.
- A. Geiszinger, W. Goessler, D. Kuehnelt, K. Francesconi and W. Kosmus, Environ. Sci. Technol., 1998, 32, 2238–2243 CrossRef CAS.
- I. Koch, I. X. Wang, C. A. Ollson, W. R. Cullen and K. J. Reimer, Environ. Sci. Technol., 2000, 34, 22–26 CrossRef CAS.
- J. S. Petrick, F. Ayala-Fierro, W. R. Cullen, D. E. Carter and H. V. Aposhian, Toxicol. Appl. Pharmacol., 2000, 163, 203–207 CrossRef CAS.
- J. S. Petrick, B. Jagadish, E. A. Mash and H. V. Aposhian, Chem. Res. Toxicol., 2001, 14, 651–656 CAS.
- T. Gebel, Chem.-Biol. Interact., 1997, 107, 131–144 Search PubMed.
- M. Vahter, R. Couch, B. Nermell and R. Nilsson, Toxicol. Appl. Pharmacol., 1995, 133, 262–268 CrossRef CAS.
- M. Vahter and E. Marafante, Arch. Toxicol., 1985, 57, 119–124 CAS.
- R. Mukhopadhyay, J. Shi and B. P. Rosen, J. Biol. Chem., 2000, 275, 21149–21157 CrossRef CAS.
- J. Y. Liu and B. P. Rosen, J. Biol. Chem., 1997, 272, 21084–21089 CrossRef CAS.
- A. A. Meharg, J. Naylor and M. R. Macnair, J. Environ. Qual., 1994, 23, 234–238 CAS.
- P. Nissen and A. A. Benson, Physiol. Plant, 1982, 54, 446–450 CAS.
- C. Langdon, A. A. Meharg, J. Feldmann, T. Balgar, J. Charnock, M. Farquhar, T. Piearce, K. Semple and J. Cotter-Howells, 5th International Conference on Environmental Biological Aspects of Main-Group Organometals (ICEBAMO), Schielleiten, 2001.
- A. Geiszinger, W. Goessler, S. N. Pedersen and K. A. Francesconi, Environ. Toxicol. Chem., 2001, 20, 2255–2262 CAS.
- I. Koch, J. Feldmann, L. X. Wang, P. Andrewes, K. J. Reimer and W. R. Cullen, Sci. Total Environ., 1999, 236, 101–117 CrossRef CAS.
- W. R. Cullen, D. Hettipathirana and J. Reglinski, Appl. Organomet. Chem., 1989, 3, 515–521 CAS.
| This journal is © The Royal Society of Chemistry 2004 |