Ligandless Stille cross-coupling in ionic liquids†
Cinzia Chiappe*a, Giovanni Imperatoa, Elio Napolitanob and Daniela Pieraccinia
aDipartimento di Bioorganica e Biofarmacia, via Bonnanno 33, Pisa, Italy. E-mail: cinziac@farm.unipi.it; Fax: +39 050 43321; Tel: +39 050 44074
bScuola Normale Superiore di Pisa, Piazza dei Cavalieri 7, Pisa, Italy. E-mail: eliona@farm.unipi.it
First published on 8th December 2003
Abstract
The Stille cross-coupling reaction has been investigated in ten different ILs to evaluate how the different physico-chemical properties of the medium can affect the transfer of vinyl and alkyl groups, as well as the efficiency of the extraction processes. The possibility of working in the absence of ligand has been also evaluated.
Introduction
Room Temperature Ionic Liquids (RTILs) are promising green alternatives to conventional solvents in transition-metal catalyzed reactions such as the Heck1 insertion reaction and Suzuki2 and Stille3 cross-coupling. The advantages of the elimination of highly toxic co-solvents, the ease of work-up (products can be isolated from the reaction mixture either by decantation or extraction with a variety of solvents, including CO2 under supercritical conditions), and the possibility of recycling the catalyst have already been demonstrated.4 Besides providing eco-friendly reaction media, one further step toward the effective and rational use of ILs in cross-coupling reactions would be an understanding of the way ILs can interact with reaction processes. In fact, ionic liquids, although sharing the important physical property of being non volatile, can exhibit quite distinct chemical properties in terms of polarity, nucleophilicity and hydrogen bonding properties,5 so that their ability to affect reaction pathways can be expected to be dependent on their structure, as is observed with common organic solvents. As a part of a general program aimed at understanding how ILs properties may affect reactivity6 and providing a rational basis for choosing among different ILs, we investigated the Stille coupling of aryl iodide with vinyl and alkyl stannanes in ten ionic liquids (Fig. 1), which have different physico-chemical properties according to the nature of the anion and cation (namely, N-butyl-N-methylimidazolium bromide [bmim][Br], hexafluorophosphate [bmim][PF6], bis(trifluoromethylsulfonyl)imide [bmim][Tf2N], tetrafluoroborate [bmim][BF4], octylsulfate [bmim][C8H17SO4], N-ethyl-N-methylimidazolium tosylate [emim][OTs], 1-butyl-2,3-dimethylimidazolium hexafluorophosphate [bm2im][PF6], bis(trifluoromethylsulfonyl)imide [bm2im][Tf2N], N-butyl-N-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide [bpyrr][Tf2N] and N-hexylpyridinium bis(trifluoromethylsulfonyl)imide [HPy][Tf2N]).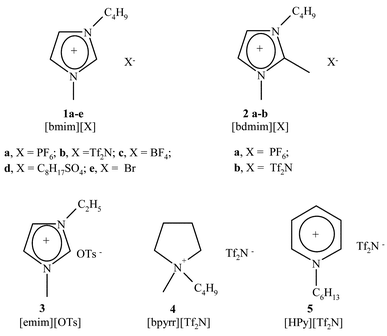 | ||
| Fig. 1 ILs used. | ||
We considered the reaction of tributylvinylstannane, tetramethylstannane and tetrabutylstannane with iodobenzene in presence or in absence of AsPh3 (“ligandless” coupling). When AsPh3 was present, Pd2(dba)3 was the source of catalyst; otherwise Pd(OAc)2 was adopted in the ligandless reaction. Although palladium catalysts rapidly dissolve in the ionic media at the reaction temperature, reagents and products are only slightly soluble, so a biphasic system resulted. In these cases cross-coupling products could, in principle, be isolated by decantation. Only in the very lipophilic [bmim][C8H17SO4] did the reaction occur in a homogeneous phase. In order to obtain comparable data, however, products were isolated in all cases by n-pentane extraction.
Results
The coupling with the vinylstannane by the Pd(0)/AsPh3 system was practically quantitative after 4 hours at 80 °C regardless of the nature of the ionic solvent (data not reported). Therefore, in order to evaluate how the solvent affected reactivity we measured the reaction progress after 1 hour (Table 1). The degree of conversion as well as the actual mass balance, which is related to the ease of extraction of reactants and products from the ionic medium, were evaluated by GC after the addition of an internal standard.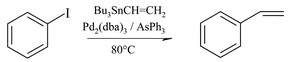
The data summarized in Table 1 show that [bmim][C8H17SO4] is the best solvent for the Stille coupling of vinylstannane and iodobenzene both in terms of yield of reaction and extraction from the reaction medium (entry 4), followed by bis(trifluoromethylsulfonyl)imide salts ([bmim][Tf2N], [HPy][Tf2N], [bpyrr][Tf2N], entries 1, 9, 10, respectively). In contrast, significantly lower yields of styrene were obtained in nucleophilic solvents like [bmim][Br] and [emim][OTs]. Modification of the structure of the cation determines appreciable variations in the coupling rate only for hexafluorophosphate derivatives (compare entries 2 and 7 with 1, 8, 9, 10). The difference seems related to a different hydrogen-bond acidity; the cation which is unable to form a hydrogen bond performs better than the other one.
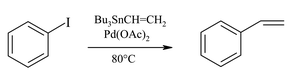
The ligandless Stille coupling was significantly less effective, as compared to the reaction with AsPh3 complexed catalyst, in all ionic liquids: up to 24 hours (as compared to 4 h) were necessary to achieve quantitative coupling. It is worth of note that under these conditions the reduction of Pd(II) to Pd(0) is immediate, therefore the reduced efficiency of the ligandless coupling cannot be attributed to this step. The data, summarized in Table 2, show that [Tf2N] derivatives give the highest reaction rates. It is worth noting that, in absence of AsPh3, the reaction seems to be more sensitive to the cation, so that ILs unable to give hydrogen bonding ensure the highest percentage of conversion (entries 8–7 vs. 1–2).
The reuse of catalyst was briefly investigated for the reaction in [bmim][C8H17SO4], [bmim][Tf2N], [bpyrr][Tf2N], and [HPy][Tf2N]: the data reveal a modest decrease in activity for Pd(II) in [bpyrr][Tf2N] (20%) and a complete loss of activity of the catalyst in the remaining ILs; the loss of catalytic activity is probably due to the precipitation of Pd(0) at the end of the extraction process.
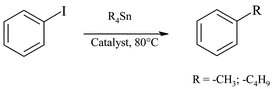
The transfer of alkyl groups from simple tetraorganotin to iodobenzene still remains difficult in the ionic medium and, in the case of methyl introduction, it is generally accompanied by the formation of biphenyl (Table 3). When Me4Sn was used as reactant and [bmim][C8H17SO4] as solvent, the introduction of the alkyl group also proceeded with the uncomplexed catalyst, although a slightly higher amount of biphenyl was detected (entries 1, 2). Once again, [bmim][C8H17SO4] and [bmim][Tf2N] are the best choice solvents for this Stille cross-coupling.
| Solvent | Catalyst | Ligand | R | Time/h | Mass balancea [%] | Ph–Rb [%] | Biphenylb [%] | |
|---|---|---|---|---|---|---|---|---|
| a Determined using benzonitrile as internal standard.b Determined by GC and GM analysis.c The unreacting material was always represented by iodobenzene. | ||||||||
| 1 | [bmim]C8H17SO4 | Pd2(dba)3 | AsPh3 | –CH3 | 24 | 100 | 31c | n.d |
| 2 | [bmim]C8H17SO4 | Pd(OAc)2 | — | –CH3 | 24 | 100 | 48c | 6 |
| 3 | [bmim]Tf2N | Pd2(dba)3 | AsPh3 | –CH3 | 24 | 90 | 62c | 27 |
| 5 | [bmim]Tf2N | Pd(OAc)2 | — | –CH3 | 24 | 70 | 12c | 30 |
| 4 | [bmim]PF6 | Pd2(dba)3 | AsPh3 | –CH3 | 24 | 87 | 54c | 28 |
| 6 | [bmim]C8H17SO4 | Pd2(dba)3 | AsPh3 | –C4H9 | 48 | 50 | 15c | <1 |
| 7 | [bmim]C8H17SO4 | Pd(OAc)2 | — | –C4H9 | 48 | 52 | 21c | n.d. |
| 8 | [bmim]Tf2N | Pd2(dba)3 | AsPh3 | –C4H9 | 48 | 70 | 60c | n.d. |
| 9 | [bmim]OTs | Pd2(dba)3 | AsPh3 | –C4H9 | 48 | 30 | 20c | n.d. |
| 10 | [bmim]Br | Pd2(dba)3 | AsPh3 | –C4H9 | 48 | 30 | 8c | n.d |
| 11 | [HPy]Tf2N | Pd2(dba)3 | AsPh3 | –C4H9 | 48 | 55 | 9c | n.d |
On the other hand, the introduction of n-butyl was extremely slow and always stopped before completion. Moreover product extraction with n-pentane was particularly ineffective as compared with previous examples. It is however worth noting that, when [bmim][Tf2N] was used as solvent, 60% of butylbenzene was obtained without the formation of the side product.
Discussion
When nucleophilic ionic liquids ([bmim][Br] and [emim][OTs]) were used as solvents in the coupling of tributylvinylstannane and iodobenzene in presence of AsPh3, only low to moderate yields of product were obtained. Non nucleophilic salts gave uniformly better results, showing that the anion identity has a great influence on the outcome of the reaction. Among all the solvents taken into account in this work, Tf2N− based ionic liquids and [bmim][C8H17SO4] gave the highest reaction rates. This could be ascribed, in the case of Tf2N− derivatives, to a nucleophilic assistance in the transmetallation step: indeed, several reports7 have recently appeared demonstrating the importance of coordinative expansion at the tin atom produced by nitrogen nucleophiles. It is worthy of note that the ability of the anion to interact with the tin atom is also affected by the nature of the cation. A competition between the cation and the Sn atom for the anion, due to the equilibria reported in Scheme 1, can be hypothesized: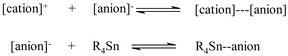 | ||
| Scheme 1 | ||
The strong cation–anion interaction present in [bmim][Br] could reduce the ability of this anion to nucleophilically assist the transmetallation and could therefore explain the low rate of the Stille cross-coupling reaction performed in this solvent, as compared1c to other Pd-mediated reaction performed in tetrabutylammonium bromide. Competing equilibria have been proposed by Welton to explain the effect of hydrogen bonding in Diels–Alder reaction and in recent polarity mesurements.8,9 Though in Diels–Alder reaction it is the ability of the cation to act as a hydrogen bond donor that determine the endo selectivity, in the Stille cross-coupling it is the hydrogen bond acceptance of the anion that can influence the result of the reaction. Therefore, it seems that both in hydrogen bond donation and acceptance, competition between the solute and the proper counterpart of the ionic liquid can be important in determining the result of a given reaction.
On the other hand, the high efficiency of the reaction in [bmim][C8H17SO4] might be related to more favorable intrinsic properties of this new ionic liquid or to the fact that reaction occurs in a homogeneous phase.
The structure of the cationic counterpart has little effect on the vinyl transfer from tetraorganotin to iodobenzene. The data reported in Table 1 show that, mantaining the anion, ionic liquids derived from different classes of organic compounds, that is imidazolium, pyridinium and pyrrolidinium salts, give almost identical results. The ability of the cation to form hydrogen bonds seems, however, to have some consequences. In particular, the introduction of a methyl group between the two nitrogen atoms in imidazolium derivatives, that should suppress the hydrogen bond acidity of the solvent, has a different effect depending on the nature of the negative counterpart: it enhances the rate of cross-coupling in the hexafluorophosphate series, whereas it has no effect on the bis(trifluoromethylsulfonyl)imide derivatives.
The data seem also to demonstrate that the reaction rates are not dependent on viscosity: comparable results are obtained for the homologous [bmim][Tf2N] and [bm2im][Tf2N], which at least at room temperature have quite different viscosities.10,11 Therefore, although the reactions occur under heterogeneous conditions, mass transfer is not the rate determining step. However, this physical property is probably more important during the isolation of the product; because of the poor material balance, the three most viscous ILs, namely [bm2im][PF6], [emim][OTs], and [bmim][Br], cannot be advantageously used as solvent for the Stille reaction (entries 5, 6, 7 from Table 1).
As shown in Table 2, ILs can effectively replace toxic solvents like HMPA in ligandless Stille cross-coupling.12 As with Pd(AsPh3)2, non nucleophilic ionic liquids having bis(trifluoromethylsulfonyl)imide as the anion are the best choice for the ligandless cross-coupling reactions (entries 1, 8, 9, 10). Apparently, when ligands are not present, the possibility of N–Sn intermolecular interactions enhances the reactivity of the system so that the coupling is complete in reasonably short times (4 hours). Furthermore, under these conditions the rate of the reaction is more sensitive to cationic structure, so that ILs incapable of forming H-bonds are preferred (compare entries 1 and 2 with 7 and 8). As in conventional solvents, the lack of coordinating ligands negatively affects the stability of the catalyst, so that recycling of the system is often difficult.
Much of the discussion of Pd catalyzed reactions in ionic liquids has focused on the possible formation of imidazolidene complexes either by deprotonation of the imidazolium cation1b,13 or by oxidative addition of the cation to the metal centre.14 As the Stille cross-coupling under investigation is sensitive to the presence of AsPh3 and the yields of conversion in [bpyrr][Tf2N] and [HPy][Tf2N] are very similar to those obtained in [bmim][Tf2N], we believe that the involvement of a metal carbene complex formed in situ is unlikely under our conditions. However, our data do not give information about the nature of the really reactive palladium species, so it is not possible to exclude the presence of Pd nanoparticles.15
Finally, although with a lower efficiency, ILs can also be used to transfer alkyl groups using simple commercial tetraalkylstannanes. Besides, the yields for methyl and butyl transfer are similar to or higher than those reported in conventional organic solvents.16 Anyway, the use of ionic liquids as the reaction media cannot be considered a variant to the use of stannatranes17 which, although not easy to synthesize, remain the reagents capable of significantly improving the alkyl transfer in Stille coupling.
Conclusion
The efficiency of Stille cross-coupling in ionic liquids is not insensitive to the nature of the ionic media. The best solvents are [bmim][C8H17SO4], [bmim][Tf2N], [bpyrr][Tf2N], and [HPy][Tf2N] either in terms of conversion or mass balance. The introduction of a vinyl group is also possible in the absence of AsPh3, even if longer reaction times are needed. On the other hand, the cross-coupling of simple tetraalkylstannanes such as Me4Sn and But4Sn with iodobenzene still remains difficult in the ionic media.Experimental
GC-MS spectra were carried out on a 30 m DB5 capillary column using an instrument equipped with an ion trap detector. GC analyses were carried out using an ECONOCAP EC-5 column (30 m). Iodobenzene, tributylvinylstannane, tetrabutylstannane and tetramethylstannane were used without purification. [bmim][PF6], [bmim][Tf2N], [bmim][Br], [bm2im][Tf2N], [bpyrr][Tf2N], [HPy][Tf2N], were prepared following the reported procedures;10,18 [bm2im][PF6], [emim][OTs], [bmim][C8H17SO4] were supplied from Solvent Innovation. The purity of ILs was checked as previously reported.6bReactions were carried out in a screw cup with Teflon-faced rubber septum vials under magnetic stirring. To a suspension of Pd2(dba)3 (0.025 mmol) and Ph3As (0.05 mmol) or Pd(OAc)2 (0.025 mmol) in 1 ml of ionic liquid were added iodobenzene (0.5 mmol) and the organostannane (0.6 mmol). The mixtures were stirred at 80 °C for the times reported in Tables 1, 2 and 3. The products were extracted with n-pentane (10 × 1 ml), the organic layers were dried over MgSO4 and diluted to an exactly known volume. A portion of this solution, exactly measured, was analyzed by GC-MS and the remaining by GC, after the addition of an appropriate amount of an internal standard (benzonitrile).
Acknowledgements
We acknowledge the financial contribution of MIUR and Università di Pisa.References
- (a) V. Calò, A. Nacci, A. Monopoli, L. Lopez and A. Di Cosmo, Tetrahedron, 2001, 57, 6071 CrossRef CAS; (b) L. Xu, W. Chen and J. Xiao, Organometallics, 2000, 19, 1123 CrossRef CAS; (c) V. P. W. Böhm and W. A. Herrmann, Chem. Eur. J., 2000, 6, 1017 CrossRef CAS.
- C. J. Mathews, P. J. Smith and T. Welton, Chem. Commun., 2000, 1249 RSC.
- S. T. Handy and X. Zhang, Org. Lett., 2001, 3(2), 233 CrossRef CAS.
- (a) H. Olivier-Bourbigou and I. Magna, J. Mol. Catal. A: Chem., 2002, 2484, 1; (b) P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772 CrossRef.
- J. L. Anderson, J. Ding, T. Welton and D. Armstrong, J. Am. Chem. Soc., 2002, 124, 14247 CrossRef CAS.
- (a) C. Chiappe, V. Conte and D. Pieraccini, Eur. J. Org. Chem., 2001, 2831; (b) C. Chiappe, D. Pieraccini and P. Saullo, J. Org. Chem., 2003, 68, 6710 CrossRef CAS.
- (a) M. T. Barros, C. D. Maycock, M. I. Madureira and M. R. Ventura, Chem. Commun., 2001, 1662 RSC; (b) J. M. Brown, M. Pearson, J. T. B. H. Jastrzebski and G. Van Koten, J. Chem. Soc., Chem. Commun., 1992, 1440 RSC; (c) V. Farina and E. Napolitano, Organometallics, 2003, 22, 4030 CrossRef.
- A. Aggarwal, N. L. Lancaster, A. R. Sethi and T. Welton, Green Chem., 2002, 4, 517 RSC.
- L. Crowhurst, P. R. Mawdsley, J. M. Perez-Arlandis, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2003, 5, 2790 RSC.
- N. L. Lancaster, P. A. Salter, T. Welton and G. B. Young, J. Org. Chem., 2002, 67, 8855 CrossRef CAS.
- S. V. Dzyuba and R. A. Bartsch, Chem. Phys. Chem., 2002, 3, 161 CrossRef CAS.
- I. P. Beletskaya, J. Organomet. Chem., 1983, 250, 551 CrossRef CAS.
- C. J. Mathews, P. J. Smith, T. Welton, A. J. P. White and D. J. Williams, Organometallics, 2001, 20, 3848 CrossRef CAS.
- P. Wasserscheid, Transition metal catalysis in ionic liquids” in Ionic liquids in synthesis, ed. P. Wassersheid and T. Welton, Wiley-VCH, 2003 Search PubMed and references therein.
- (a) V. Calò, A. Nacci, A. Monopoli, S. Laera and N. Cioffi, J. Org. Chem., 2003, 68, 2929 CrossRef CAS; (b) M. T. Reetz and M. Masse, Adv. Mater., 1999, 11, 773 CrossRef CAS.
- (a) V. Farina, S. Kapadia, B. Krishnan, C. Woung and L. S. Liebeskind, J. Org. Chem., 1994, 59, 5905 CrossRef CAS; (b) F. Bellina, A. Carpita, D. Ciucci, M. De Santis and R. Rossi, Tetrahedron, 1993, 49, 4677 CrossRef CAS; (c) D. Milistein and J. K Stille, J. Am. Chem. Soc., 1979, 101, 4993.
- E. Vedejs, A. R. Haight and W. O. Moss, J. Am. Chem. Soc., 1992, 114, 6556 CrossRef CAS.
- (a) R. T. Fuller, H. C. Carlin, D. De Long and Haworth, J. Chem. Soc., Chem. Commun., 1994, 299 RSC; (b) L. Cammarata, S. G. Kazarian, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2001, 3, 5192 RSC; C. Chiappe and D. Pieraccini, Green Chem., 2003, 5, 193 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Stille coupling of iodobenzene with tributylvinylstannane in ionic liquids with complexed palladium catalyst. See http://www.rsc.org/suppdata/gc/b3/b313221h/ |
| This journal is © The Royal Society of Chemistry 2004 |