Structure–reactivity correlations in the selective aerobic oxidation of cinnamyl alcohol: in situ XAFS
Adam F. Lee* and Karen Wilson
Department of Chemistry, University of York, York, UK YO10 5DD. E-mail: afl2@york.ac.uk; Fax: +44 1904 432516; Tel: +44 1904 434470
First published on 21st November 2003
Abstract
The structural evolution of a Pd/C catalyst during the liquid phase selective aerobic oxidation of cinnamyl alcohol has been followed by in situ XAFS and XPS. The fresh catalyst comprised highly dispersed, heavily oxidised Pd particles. Cinnamyl alcohol oxidation resulted in the rapid reduction of surface palladium oxide and a small degree of concomitant particle growth. These structural changes coincided with a large drop in catalytic activity. Prereduced Pd/C exhibited a significantly lower initial oxidation rate demonstrating the importance of surface metal oxide in effecting catalytic oxidation. Use of a Pd black model system confirmed that the oxide→metal transformation was the cause, and not result, of catalyst deactivation.
Green ContextThe selective oxidation of organic compounds is probably the most widely used synthetic transformation in chemistry. Catalytic aerobic oxidation is normally regarded as the best green chemistry option but such systems are generally poorly understood. Here the structural evolution of a typical oxidation catalyst, supported palladium, during an aerobic oxidation is studied using in situ XAFS. The study proves to be viable and proves the importance of surface oxide sites. The better understanding of such procedures should help us to develop and apply many more green oxidation systems based on heterogeneous catalysts.JHC |
Introduction
The selective oxidation of alcohols finds application in the synthesis of many valuable intermediates for the food and pharmaceutical industry. Supported platinum-group metals have long shown promise in the aerobic partial oxidation of carbohydrates and alcohols1 over wide temperature, pressure and solvent regimes and have been extensively studied over the past decade.2 Bimetallic variants incorporating a second inert component have also been shown to enhance the selectivity of such catalysts though the origin of such promotional effects remains contentious. Despite their potential as generic selective oxidation catalysts, further development is hindered by the complexity of the three-phase system, and severe deactivation of these materials. Two issues in particular require addressing; the nature of the active site and the deactivation mechanism.Three main sources of catalyst deactivation have been advanced in the literature for the liquid phase oxidation of alcohols. First, so called chemical poisoning, i.e. the irreversible adsorption of strongly bound hydrocarbons, including reaction intermediates and (by) products such as carboxylic acids. This is commonly reported in aqueous media working at low pH3 or over reduced metal surfaces (low catalyst potential),4 although secondary alcohols are reported as insensitive to pH.5 Despite many electrochemical studies of related poisoning phenomena6–8 there are no direct measurements on the nature or binding strength of such adsorbates over dispersed noble metal catalysts. Surface restructuring and corrosion may also contribute to catalyst deactivation, most notably for promoted catalysts wherein the inactive component (usually Bi, Pb or Sn) is readily leached into aqueous solution.9 This is most problematic when reactions are performed in the presence of chelating agents or at acidic pH.3,10–12 Under aqueous conditions leaching is minimised through the use of a Na2CO3 or Li2CO3 buffer to maintain pH > 8. However the most important deactivation process relates to the oxidation state of the catalyst surface. From electrochemical measurements, it is widely held that alcohol oxidation occurs over metal (Pt0) sites2 and that poor rates are observed over heavily oxidised catalysts.13,14 However inefficient reactant diffusion under these simulated reaction conditions and the inherent invasive nature of electrochemical measurements may result in perturbation of the chemical state of the catalyst surface. Thus despite this common assertion there remain no direct structural investigations of the working catalyst during reaction. Consequently, in the absence of oxygen mass-transport limitations, it has been proposed that over-oxidation causes the rapid deactivation of catalysts with intrinsically poor activity.15 Indeed it has been suggested that bulk platinum oxides13 or a platinum-oxygen solid solution14,15 can form under these mild reaction conditions. In contrast Baiker and coworkers suggest that over-oxidation is the result (not cause) of chemical poisoning, noting that a partial oxygen coverage is essential for preserving high activity.16 Indeed cinnamyl alcohol oxidation is greatly suppressed over reduced Pt,Bi/Al2O3 in comparison with its oxidised counterpart.17 Clearly a subtle interplay exists between competitive adsorption of organic moieties and oxygen.
Identification of the reaction-induced morphological and chemical changes from these early studies are also complicated by the use of aqueous reaction media necessitating surfactants and pH regulation via base addition; the latter may induce oscillations in the steady state oxidation rate.18 Utilisation of aqueous solvents is often justified on the grounds that for safety reasons industrial scale oxidation reactions cannot be performed in hydrocarbon solvents and that water is the ideal ‘green’ solvent. However there are several problems associated with the use of water, namely that many organic compounds require the use of surfactants to solubilise them, increasing the processing steps and associated solvent waste produced on extraction of the product. Additionally the aqueous waste stream contaminated with any soluble organic components is particularly difficult to treat.19 Thus the environmental impact of both treating the waste water, and energy input to separate the surfactant from the product versus distillation and recycling of a benign organic solvent, must be considered.
In situ XAFS has emerged as a powerful technique for studies of catalyst evolution during activation treatments20 or steady state measurements of catalyst oxidation.21 Previously we have shown that allylic alcohol oxidation can be performed effectively in an organic solvent (ethanol, toluene),22 vastly simplifying catalytic mechanistic studies. Here we address the role of metal oxidation state in cinnamyl alcohol oxidation in a hydrocarbon solvent via spectroscopic measurements of a monometallic Pd/C catalyst. By omitting promoters we avoid issues concerning their disruption of the active ensemble through surface decoration or alloying. Complications arising from modified oxygen/substrate activation and associated spillover processes are also eliminated. Likewise use of a hydrocarbon solvent eliminates the necessity for surfactant, and improves diffusion of organic reactant. Specifically we build upon our original report23 of the application of in situ XAFS to follow the real-time, dynamic structural evolution of a heterogeneous catalyst during a liquid phase organic reaction. A transformation from oxidic to metallic Pd particles precisely correlates with severe catalyst deactivation demonstrating the importance of surface oxides in selective oxidation.
Experimental
In situ XAFS measurements were made on Station 9.2 of the Daresbury SRS facility, using a Si(311) double-crystal monochromator with a beam current/energy of 150 mA/2 GeV. Pd (24.35 keV) Quick XAFS spectra were acquired in a specially constructed, aluminium recirculating trickle-bed reactor based on an existing design.21,24 Approximately 0.1 g of 5 wt% Pd/charcoal catalyst powder was diluted with 0.7 g of boron nitride and loaded into the cell, supported on a silica wool bed between 25 µm Kapton windows giving an overall X-ray path length of 13 mm. The reaction mixture, comprising 5 mmol cinnamyl alcohol (Aldrich 99%) in 60 ml toluene, equivalent to a 1 : 112 molar ratio, was fed from an oxygen-saturated external reservoir at ∼20 ml min−1. Before reaction, both the reservoir and catalyst were heated to 60 °C before circulating the reaction mixture. XAS spectra were taken every 3 minutes for a 10 h period after which cinnamyl alcohol conversion reached a plateau. Spectra were fitted using the Daresbury ECABS and EXCURV98 packages for background subtraction, and phaseshift determination and fitting procedures respectively. Reference transmission XAFS of PdO (>99% Lancaster) and a 10 µm Pd foil standard were also recorded.An inert internal standard, mesitylene (Aldrich 99%) was included for calculation of product yields and to ensure closure of the carbon mass balance (>95%). Aliquots (0.1 ml) of the reaction mixture were periodically withdrawn for off-line analysis using a Perkin-Elmer 8500 GC and a 30 m × 0.25 mm HP5 capillary column. Cinnamaldehyde was the principal reaction product, with a small amount of phenylpropanol and trace cinnamic acid also formed. There were no side-products attributable to solvent oxidation. Quoted conversions are ± 1% and selectivities ± 3%. Cinnamyl alcohol oxidation was also followed in situ by monitoring the oxygen uptake from the reservoir deadspace (maintained at 1 bar O2) using a Buchi Pressflow Gas Controller.
X-Ray photoelectron spectra were acquired using a Kratos Axis HSi instrument operating at 225 W with a Mg Kα excitation source (1253.6 eV) and a CHA analyser selecting at 20 eV pass energy. The sample was charge neutralised and Pd 3d, and C and O 1s spectra recorded at normal emission to the surface. Binding energies were referenced to the C 1s of graphite at 285 eV. X-Ray diffractograms were recorded on a Siemens D5000 diffractometer using Cu Kα radiation and a 2θ scan range of 10–105° in 0.02° steps. Surface areas were determined by N2 physisorption and metal dispersions by CO chemisorption.
Results and discussion
X-Ray photoelectron spectroscopy
Changes in the surface oxidation state during cinnamyl alcohol oxidation were determined from Pd XP spectra of the fresh and spent Pd/charcoal catalysts (Fig. 1). The Pd 3d spectra show that the fresh, untreated catalyst contains two palladium states consistent with metallic and oxidic (2+) environments,25 the 3d5/2 components at 335.5 eV and 337.3 eV binding energy (BE) respectively. Approximately 70% of the total surface palladium is initially present as oxide, indicative of small particles (<50 Å)26,27 in accordance with surface energy constraints.28Fig. 1 shows that catalytic oxidation of a 5 mmol cinnamyl alcohol mixture under mild reaction conditions dramatically reduces the amount of surface PdOx to below 12%. The total Pd surface concentration is unchanged during reaction, consistent with ICP analysis of hot filtered, post-reaction catalysts which reveal negligible metal leaching. The corresponding O 1s XP spectra (not shown) reveal a 55% decrease in surface oxygen confirming the reduction of surface Pd during cinnamyl alcohol oxidation. Fig. 1 also shows that in situ chemical reduction yields a similar spectrum to that obtained from Pd/C directly reduced via H2 at 190 °C. The stoichiometry of the initial surface oxide can be calculated from the relative Pdδ+ 3d : O 1s peak intensities and taking into account the appropriate correction factors yields a 1 : 1 ratio consistent with the presence of PdO.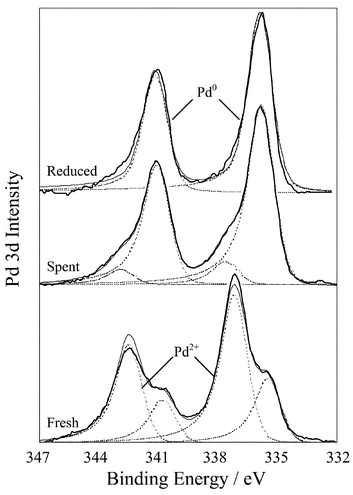 | ||
| Fig. 1 Pd 3d XP spectra of fresh and spent Pd/C catalyst after 10 h cinnamyl alcohol oxidation. A spectrum of the H2 reduced catalyst is shown for comparison. | ||
In situ EXAFS
In order to follow the dynamics of this reduction process Pd K-edge EXAFS were acquired in situ during cinnamyl alcohol oxidation, Fig. 2. Measurements of the fresh catalyst within the reaction cell confirm the presence of both Pd oxide and metal environments. Fig. 3a and b show the respective raw χ data and radial-distribution function prior to reaction: Pd–O and Pd–Pd coordination shells (r) are present at ∼2.03 and 2.75 Å respectively. The low coordination numbers (CN) shown in Table 1 are consistent with small (<20 Å) Pd particles capped by a truncated, surface oxide phase. The alternative possibility, that the catalyst comprises a mixture of discrete oxide and metal crystallites is unsupported by XRD.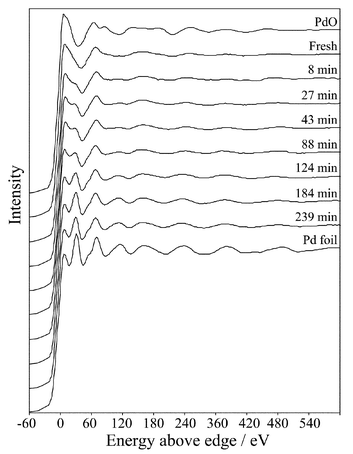 | ||
| Fig. 2 Time-resolved Pd K-edge quick XAFS of a Pd/C catalyst during cinnamyl alcohol oxidation. Reference spectra of PdO and a Pd foil are shown for comparison. | ||
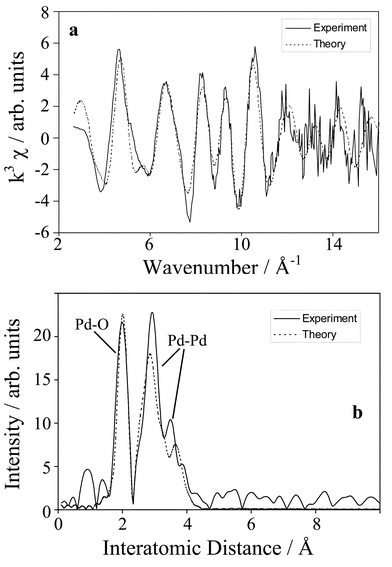 | ||
| Fig. 3 Pd K-edge (a) k3-weighted raw EXAFS and (b) pseudo-radial distribution function of a fresh Pd/C catalyst. | ||
| Parameter | PdO | Fresh | Spent | Pd Foil |
|---|---|---|---|---|
| CN1Pd–O | 4 | 2.5 | — | — |
| CN1Pd–Pd | 8 | 1.7 | 4.45 | 12 |
| CN2Pd–Pd | 2 | 2.8 | — | 6 |
| r1Pd–O / Å | 2.03 | 2.04 | — | — |
| r1Pd–Pd / Å | 3.07 | 2.76 | 2.76 | 2.74 |
| r2Pd–Pd / Å | 3.45 | 3.43 | 3.87 | |
| σ1Pd–O | 0.007 | 0.007 | — | — |
| σ1Pd–Pd | 0.017 | 0.012 | 0.011 | 0.014 |
| σ2Pd–Pd | 0.005 | 0.018 | — | 0.02 |
The Pd K-edge spectra remain unchanged for ∼40 min following introduction of the reaction medium to the catalyst, Fig. 4, a period during which cinnamyl alcohol oxidation proceeds with a constant (maximum) rate. Subsequently the white line intensity (edge-jump), a direct measure of the Pd oxidation state,29 shows a sharp drop which is complete after 120 min reaction, associated with a transformation from oxidic to metallic palladium.
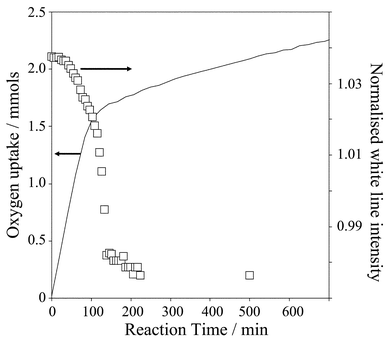 | ||
| Fig. 4 Correlation between oxygen uptake and normalised Pd K-edge white line intensity as a function of reaction time. | ||
The fitted χ data and radial-distribution function (Fig. 5a and b) confirm the loss of the Pd–O coordination shell and concomitant increased Pd–Pd scattering consistent with pure Pd particles. This structural evolution coincides with a rapid decrease in catalyst activity; the oxidation rate falls from ∼0.18 to 0.02 mmol min−1 (g cat)−1. Loss of activity due to particle sintering upon PdO reduction can be discounted as XAFS provides both oxidation state and particle size information, and reveals that the latter remains unchanged over this time period. Following this chemical reduction the average Pd coordination number begins to rise giving a limiting particle size of ∼80 Å (confirmed by XRD and TEM measurements). In all cases the Debye–Waller factors (σ), a measure of vibrational disorder, were low and in good agreement with the respective Pd oxide or metal standards. Of course it is important to establish whether these values represent true catalytic rates [eqn. (1)] or simply reflect the stoichiometric consumption of PdO [eqn. (2)].
Ph–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CH2OH +
½ O2
→ Ph–CHCH–CHO + H2O CH–CH2OH +
½ O2
→ Ph–CHCH–CHO + H2O | (1) |
| Ph–CHCH–CH2OH + PdO → Ph–CHCH–CHO + Pd + H2O | (2) |
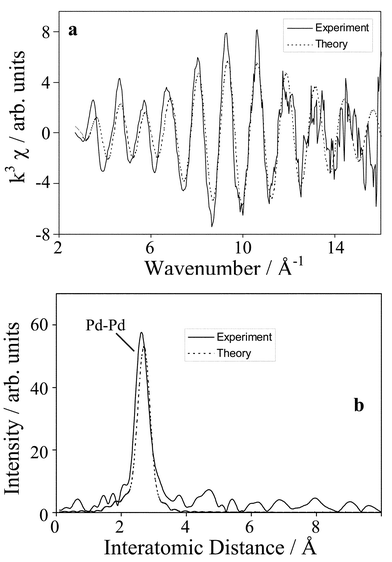 | ||
| Fig. 5 Pd K-edge (a) k3-weighted raw EXAFS and (b) pseudo radial distribution functions of a spent Pd/C catalyst. | ||
From XPS the fresh catalyst contains ∼2.86 × 10−5 moles of PdO. Assuming a 1 : 1 stoichiometry for the reaction between Oa, liberated by PdO decomposition, and cinnamyl alcohol to form cinnamaldehyde, and that catalyst reduction is complete after 120 minutes, a maximum rate of ∼0.0024 mmol min−1 (g cat)−1 is obtained. This value is much less than the measured rates and we can thus neglect direct contributions from the catalyst reduction.
Bulk catalyst properties
Gross morphological changes accompanying oxidation were examined ex situ by TEM and X-ray diffraction of the fresh and post-reaction Pd/C catalysts. Both techniques gave an average initial Pd particle size of ∼20 Å, consistent with the EXAFS estimate30,31 and the high dispersion (dPd = 1.02) indicated from CO titration. Following reaction the particle size increased to ∼100 Å in accordance with the stronger XAFS oscillations. A similar increase was observed following H2 pretreatment at 190 °C.Reactor studies
The preceding structural measurements showed the loss of surface Pd oxide during the initial stages of cinnamyl alcohol oxidation, with the limiting performance of a fresh Pd/C catalyst of ∼98% conversion and ∼90% selectivity towards the aldehyde after ∼10 h. In order to explore whether catalyst reduction was a cause of the rapid deactivation or simply a side-effect, a fresh catalyst was prereduced under H2 (190 °C) prior to reaction. The resulting catalyst was transferred under nitrogen to the trickle-bed reactor and monitored via on-line oxygen consumption and GC sampling. Fig. 6 compares the activity of the fresh and prereduced catalysts over the course of reaction. It is immediately apparent that the initial oxidation rate is much slower over prereduced Pd/C at only ∼0.11 mmol min−1 (g cat)−1. The reduced catalyst undergoes somewhat slower deactivation than fresh Pd/C but attains a similar low steady state reaction rate consistent with a common (metallic) surface environment. This limiting rate is likely controlled by the accumulation of irreversibly bound (by) products which strongly chemisorb to metal sites.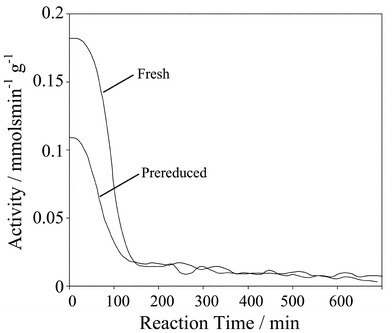 | ||
| Fig. 6 Comparison of cinnamyl alcohol oxidation rates over fresh and prereduced catalysts. | ||
Model catalyst studies
The hypothesis that metal oxide sites play an essential role in cinnamyl alcohol oxidation over Pd/C was tested using Pd black as a model catalyst to eliminate metal-support interactions and spillover effects. The fresh Pd black sample comprised large particles with a volume-averaged particle diameter of 180 Å from XRD (Fig. 7). The total surface area of fresh Pd black was only ∼30 m2 g−1 corresponding to a metal dispersion of only 0.08. Palladium 3d XP spectra in Fig. 8 show that the fresh catalyst contains ∼33% oxide, the greater proportion of metallic palladium reflecting the much lower dispersion compared to the Pd/C catalyst.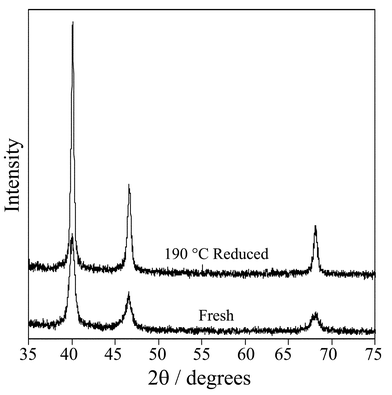 | ||
| Fig. 7 X-Ray diffractograms of fresh and 190 °C H2 reduced Pd black. | ||
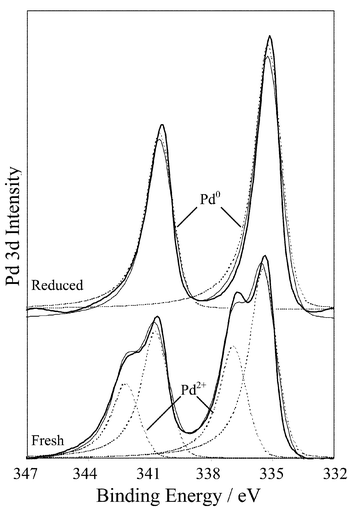 | ||
| Fig. 8 Pd 3d XP spectra of fresh and prereduced Pd black. | ||
Direct low temperature (<200 °C) reduction under H2 was subsequently attempted. As expected reduced Pd black samples transferred under nitrogen showed a significant increase in average particle size determined by XRD, reaching ∼600 Å after 190 °C reduction. The associated Pd 3d XP spectra reveal a dramatic change in surface composition with only metallic Pd sites remaining following reduction. Fig. 9 compares the catalytic performance of fresh and reduced Pd black samples towards cinnamyl alcohol oxidation. Conversion over fresh Pd black was significantly lower (∼19%) than that observed over Pd/C under identical reaction conditions reflecting the great difference in overall metal dispersion. Reduction temperatures as low as 130 °C proved sufficient to dramatically decrease catalytic activity, which was completely suppressed above 190 °C.
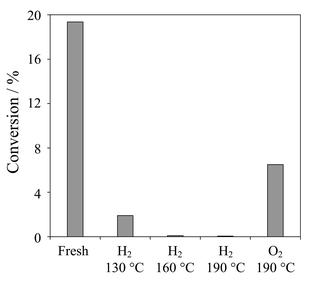 | ||
| Fig. 9 Effect of catalyst pretreatment on cinnamyl alcohol conversion over fresh, reduced and reoxidised Pd black after 10 h reaction. | ||
This transformation correlates directly with the loss of active surface palladium oxide (and not particle size effects). Since reduced samples were cooled under nitrogen any contribution from a palladium hydride phase may also be discounted. Confirmation for the crucial role of surface oxide in cinnamyl alcohol reaction was obtained from the reoxidation of fully reduced Pd black; calcination under oxygen (10 ml min−1 at 190 °C for 2 h) resulted in partial regeneration of the original oxidation performance. A similar effect was observed following ambient addition of 40 w/v% H2O2 solution to reduced Pd black followed by filtration and drying. These results also demonstrate that poisoning due to surface reduction alone is reversible. In contrast pure PdO particles showed no activity towards cinnamyl alcohol presumably reflecting the high thermodynamic stability of bulk versus surface oxides.
In situ XAFS has enabled us to identify the reduction of catalytically active PdOx surface sites during Pd/C catalysed allylic alcohol oxidation. That this proposed deactivation mechanism differs from reactions performed in aqueous media may be ascribed to the relative solubilities of the reactants. Operation in aqueous solvents, wherein allylic alcohol solubilities and diffusion rates are so poor as to necessitate surfactant addition, result in high O2 : substrate ratios which thereby favour catalyst overoxidation. However in organic media, cinnamyl alcohol diffusion to the catalyst surface is facile, preventing overoxidation. The predominant catalyst deactivation route now occurs via reduction of surface PdO→Pd and associated build-up of irreversibly adsorbed by-products on the resultant metal surface. Recent ATR measurements support that under these conditions catalyst deactivation occurs via site blocking by adsorbed CO32 formed during decarbonylation reactions.
Conclusions
In situ XAS has been shown to be a viable tool for following structural changes within working supported heterogeneous catalysts during liquid phase reactions. The rate of cinnamyl alcohol oxidation to cinnamaldehyde directly correlates with the oxidation state of highly dispersed Pd particles. A surface palladium oxide layer is essential for maintaining high selective oxidation activity. Adsorbate-induced catalyst reduction and restructuring (prevalent under oxygen mass-transfer limited conditions), dramatically lowers catalyst activity possibly associated with the rapid deactivation of Pd metal versus oxide surface sites.Acknowledgements
Financial support by the UK EPSRC under grant GR/M20877 and the UK CLRC under grant 37379 is gratefully acknowledged. We also thank Johnson Matthey plc for the supply of precious metals.References
- A. Strecker, Ann., 1855, 93, 370 Search PubMed; H. Weiland, Ber., 1921, 54, 2353 Search PubMed.
- T. Mallat and A. Baiker, Catal. Today, 1994, 19, 247 CrossRef CAS; M. Besson and P. Gallezot, Catal. Today, 2000, 57, 127 CrossRef; R. Sheldon, I. W. C. E. Arends and A. Dijksman, Catal. Today, 2000, 57, 157 CrossRef CAS.
- P. Fordham, M. Besson and P. Gallezot, Catal. Lett., 1997, 46, 195 CrossRef CAS.
- A. Abbadi, M. Makkee, W. Visscher, J. A. R. Vanveen and H. Vanbekkum, J. Carbohydr. Chem., 1993, 12, 573 CAS.
- J. W. Nicoletti and G. M. Whitesides, J. Phys. Chem., 1989, 93, 759 CrossRef CAS.
- J. F. E. Gootzen, A. H. Wonders, A. P. Cox, W. Visscher and J. A. R. vanVeen, J. Mol. Catal. A., 1997, 127, 113 CrossRef CAS.
- A. P. Markusse, B. F. M. Kuster and J. C. Schouten, J. Mol. Catal. A., 2000, 158, 215 CrossRef CAS.
- T. Mallat, Z. Bodnar, P. Hug and A. Baiker, J. Catal., 1995, 153, 131 CrossRef CAS.
- T. Mallat, C. Bronnimann and A. Baiker, Appl. Catal. A, 1997, 149, 103 CrossRef CAS.
- C. Bronnimann, Z. Bodnar, P. Hug, T. Mallat and A. Baiker, J. Catal., 1994, 150, 199 CrossRef.
- Y. Schuurman, B. F. M. Kuster, K. Vanderwiele and G. B. Marin, Appl. Catal. A, 1992, 89, 47 CrossRef CAS.
- M. Besson and P. Gallezot, Catal. Today, 2000, 57, 127 CrossRef.
- J. M. H. Dirkx and H. S. Vanderbaan, J. Catal., 1981, 67, 1 CAS; J. M. H. Dirkx and H. S. Vanderbaan, J. Catal., 1981, 67, 14 CAS.
- P. J. M. Dijkgraaf, H. A. M. Duisters, B. F. M. Kuster and K. Vanderwiele, J. Catal., 1988, 112, 337 CAS.
- J. H. J. Kluytmans, A. P. Markusse, B. F. M. Kuster, G. B. Marin and J. C. Schouten, Catal. Today, 2000, 57, 143 CrossRef.
- T. Mallat, Z. Bodnar and A. Baiker, J. Catal., 1995, 153, 131 CrossRef CAS.
- T. Mallat and A. Baiker, Top. Catal., 1999, 8, 115 CrossRef CAS.
- L. Jelemensky, B. F. M. Kuster and G. Marin, Catal. Lett., 1995, 30, 269 CrossRef.
- T. Y. Zhang, Waste Minimisation in Pharmaceutical Process Development: Principles, Practice & Challenges, Chapter 12, in Handbook of Green Chemistry & Technology eds. J. H. Clark and D. J. Macquarrie, Blackwell Publishing Ltd, Oxford, 2002 Search PubMed.
- J. M. Thomas and G. N. Greaves, Science, 1994, 265, 1675 CAS.
- A. P. Markusse, B. F. M. Kuster, D. C. Koningsberger and G. B. Marin, Catal. Lett., 1998, 55, 141 CrossRef CAS.
- A. F. Lee, J. J. Gee and H. J. Theyers, Green Chem., 2000, 2, 279 RSC.
- A. F. Lee, Abstr. Pap. Am. Chem. Soc., 2001, 221 Search PubMed , 187-COLL Part 1.
- H. H. C. M. Pinxt, B. F. M. Kuster, D. C. Koningsberger and G. B. Marin, Catal. Today, 1998, 39, 351 CrossRef CAS.
- C. D. Wagner, A. V. Naumkin, A. Kraut-Vass, J. W. Allison, C. J. Powell, J. R. Rumble Jr., NIST Standard Reference Database 20, Version 3.2 2000 Search PubMed.
- M. Lyubovsky, L. Pfefferle, A. Datye, J. Bravo and T. Neslon, J. Catal., 1999, 187, 275 CrossRef CAS.
- C. B. Wang and C. T. Yeh, Appl. Catal. A, 2001, 209, 1 CrossRef CAS.
- C. T. Campbell, Surf. Sci. Rep., 1997, 27, 1 CrossRef.
- T. K. Sham, S. J. Naftel and I. Coulthard, J. Appl. Phys., 1996, 79, 7134 CrossRef CAS.
- B. S. Clausen, L. Grabaek, H. Topsoe, L. B. Hansen, P. Stoltze, J. K. Norskov and O. H. Nielsen, J. Catal., 1993, 141, 368 CrossRef CAS.
- A. Jentys, Phys. Chem. Chem. Phys., 1999, 1, 4059 RSC.
- C. Keresszegi, T. Burgi, T. Mallat and A. Baiker, J. Catal., 2002, 211, 244 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2004 |