Chitosan-based heterogeneous catalysts for Suzuki and Heck reactions
Jeff J. E.
Hardy
a,
Sandrine
Hubert
a,
Duncan J
Macquarrie
*a and
Ashley J
Wilson
b
aDepartment of Chemistry, University of York, Heslington, York, UK YO10 5DD. E-mail: djm13@york.ac.uk; Fax: 0044 1904 434550; Tel: 0044 1904 434533
bDepartment of Biology, University of York, Heslington, York, UK YO10 5DD
First published on 28th November 2003
Abstract
Novel supported palladium catalysts have been developed based on chitosan as a support. These catalysts display excellent activity in the Suzuki and Heck reactions.
Introduction
The development of heterogeneous catalysts for fine chemicals synthesis has become a major area of research recently, as the potential advantages of these materials (simplified recovery and reusability, the potential for incorporation in continuous reactors and microreactors) over homogeneous systems can make a major impact on the environmental performance of a synthesis.1,2 The majority of these novel catalysts are based on silicas, primarily since silica displays many advantageous properties – excellent stability (chemical and thermal), high surface area, good accessibility, and organic groups can be robustly anchored to the surface, to provide catalytic centres. However, it does have drawbacks – it has limited stability in aqueous – especially basic – conditions, and it cannot easily be formed into membranes or into other forms which could be attached to novel reactors for use in intensive processing applications.Chitosan is produced by the deacetylation of chitin, a major naturally occurring biopolymer, which is one of the key constituents of the shells of crustaceans, and is a by-product of the fishing industry. Its structure is shown in Fig. 1. It has many applications as an adsorbent of metals, and also in medicine, where it is used in several applications, including wound dressings/artificial skin, in drug delivery and contact lenses, amongst others and is readily formed into films or fibres for many of these applications.3 The flexibility of the material, insoluble in the vast majority of solvents, but capable of being cast into films and fibres from dilute acid, along with its inherent chirality, makes chitosan an excellent candidate as a support for catalysis. In this respect, several groups have demonstrated the catalytic activity of salts physisorbed onto chitosan and reduced to the metal for the reduction of (a) chromate (using Pd),4 (b) phenol (Pd),5–7 and (c) nitro-aromatics (Ni, Cu, Cr, Zn).8 Asymmetric hydrogenation has been described by Yuan et al.9 Quignard et al. have utilised the hydrophilic nature of chitosan to prepare supported aqueous phase (SAP) catalysts for the Pd-catalysed allylic substitution reaction.10,11 Functionalisation of the chitosan to provide co-ordination sites has also been carried out and has provided catalysts for cyclopropanation of olefins (Cu-Schiff's base),12 oxidation of alkylbenzenes (Mn or Ni-Schiff's base),13 and the oxidation of DOPA (3,4-dihydroxyphenylalanine)–(Co-salen).14 The latter example involved the dissolution of chitosan in acetic acid, functionalisation and re-precipitation, whereas the others involved the direct functionalisation of the solid chitosan.
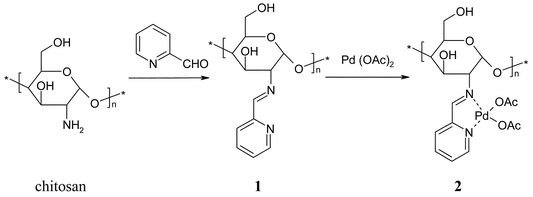 | ||
| Fig. 1 The synthesis of chitosan-pyridyl imine complex and its conversion to the related Pd complex. | ||
We now present our work on the catalytic activity of Pd-iminopyridyl complexes supported on chitosan for the Suzuki and Heck reactions.
Experimental
All chemicals were purchased from Aldrich or Lancaster, and used without further purification. Chitosan was provided by Prof. A. J. Wilson of the Department of Biology, University of York in a powdered form.Elemental analysis was provided by the University of Newcastle (combustion analysis for CHN and ICP analysis for Pd). Samples for Pd analysis were stirred with 10% HCl twice (24 h each) and the liquids combined to provide the ICP samples.
Modification of chitosan
Chitosan (1.0 g, equivalent to 6 mmol NH2) and 2-pyridinecarboxaldehyde (0.643 g, 6 mmol) were added to ethanol (50 mL) and refluxed for 18 h. The resultant mixture was cooled and the solid filtered, washed with ethanol and dried under vacuum for 6 h to give 1.1 (1.0 g) was then treated with a solution of palladium acetate (0.112 g, 0.5 mmol) in acetone (50 mL). After adsorption of the palladium, the solid was preconditioned by washing thoroughly to remove any loose Pd species. The washing consisted of three 3 h refluxes in each of ethanol, toluene, and acetonitrile. Finally the catalyst was dried at 90 °C for 18 h to give the catalyst 2.
Typical Suzuki reaction
A quantity of 2 (see Fig. 1, Results and discussion) was suspended in o-xylene (20 mL) and benzene boronic acid (0.914 g, 7.5 mmol), bromobenzene (0.805 g, 5.1 mmol), potassium carbonate (1.382 g, 10 mmol) and n-dodecane (0.749 g, 4.4 mmol) were added. The reaction was then heated at the appropriate temperature and followed by GC using n-dodecane as internal standard.Typical Heck reaction
2 (25 mg) was suspended in anhydrous dioxane (30 mL). To this were added iodobenzene (1.03 g, 5.3 mmol), n-butyl acrylate (0.68 g, 5.3 mmol) and triethylamine (0.98 g, 10 mmol). The mixture was refluxed and followed by GC, using n-dodecane as internal standard. In a second reaction, styrene and bromobenzene were coupled.Results and discussion
Preparation and analysis of catalyst
CHN analysis of the chitosan indicated, as expected, the presence of N-acetyl groups due to incomplete hydrolysis of the chitin precursor. From the analysis, approximately 35% of the nitrogen sites are acetylated, leaving a total of 3.7 mmol g−1 present as free NH2 units. Of these, 38% appear to have been functionalised with pyridylimine groups during the formation of 1. Analysis after addition of palladium acetate and thorough preconditioning by multiple washing indicates that some of these groups are lost during the preconditioning step, and the final loading of the pyridylimine group drops to 25%, corresponding to a loading of 0.92 mmol g−1 of ligand sites. The Pd content of the final catalyst was measured before and after preconditioning. Before this step, the content was 0.22 mmol g−1, while after the washing steps, this had dropped to 0.16 mmol g−1. Thus, the final catalyst has approximately 18% of the ligands occupied by metal.This low loading may reflect that many of the sites in chitosan are inaccessible to the ligand or to the Pd source during preparation. Given that chitosan is a very weakly swelling polymer, this is likely to be a reasonable explanation, although no attempts have been made to increase this figure by e.g. extended reaction times. Our previous experience with Pd catalysts has indicated that low metal loadings do not necessarily detract from activity.15 Others have reached similar conclusions.
The preconditioning steps have been previously developed to avoid leaching on silica-based analogous catalysts, by removing loosely bound Pd;15 we and others have noted that leaching does occur with these silica materials during reaction when this step is not carried out.15,16
Infra-red analyses of the chitosan catalysts were carried out using Diffuse Reflectance IR. The spectrum of chitosan has a strong and broad OH absorption centred around 3500 cm−1 which masks the N–H str. A diagnostic peak appears on condensation of the pyridine-aldehyde with the amine group at 1638 cm−1 corresponding to the formation of the pyridylimine unit. No H-bonded or free aldehyde were evident in the range 1675–1710 cm−1. Complexation of the palladium led to minor changes in the IR spectrum, as expected from previous work using this ligand system supported on silica, and from the relatively low degree of adsorption of Pd which leaves most of the imine sites unaltered.
Initial runs in the Suzuki reaction were carried out using 100 mg of catalyst (to 5.1 mmol substrate) at 130 °C. With catalyst 2 this led to a 48% yield of biphenyl after 1 h and 55% after 6 hours. Thus the reaction proceeds successfully with 2 and is almost complete after 1 h. Variation of the amount of catalyst was investigated, with the results being displayed in Fig. 2. It was found that the optimum amount of catalyst corresponded to a substrate ∶ Pd molar ratio of 1200, with no significant benefit in larger amounts of catalyst, and a significant tail-off at lower quantities. Runs without benzene boronic acid or without bromobenzene failed to produce any biphenyl, indicating the absence of any homo-coupling in the reaction.
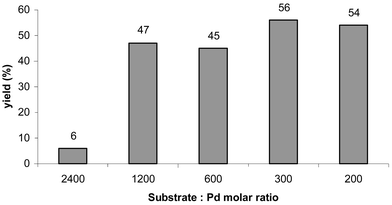 | ||
| Fig. 2 Effect of substrate ∶ Pd ratio on yield of biphenyl at 130 °C. | ||
An analogous catalyst based on salicylaldehyde (instead of the pyridyl aldehyde) was prepared, but gave poor results. Salicylaldehyde-based ligands on silicas also gave poor results.15,17
Increasing the reaction temperature to reflux (143 °C) improved the yield significantly (Table 1), with lower temperatures leading to lower conversions. All further reactions were therefore carried out at this higher temperature.
| Temperature/°C | % Biphenyla | % PhBr remaininga |
|---|---|---|
| a After 1 h (figures in brackets denote conversions after 6 h). b Catalyst reused directly. c Catalyst from run (a) reused after washing with methanol. d Catalyst after 5 uses. | ||
| 75 | 11 (13) | 84 (85) |
| 100 | 25 (29) | 72 (70) |
| 130 | 48 (55) | 49 (46) |
| 143 | 80 (87) | 17 (13) |
| 143b | 53 (57) | 50 (45) |
| 143c | 78 (86) | 20 (13) |
| 143d | 74 (81) | 22 (15) |
Catalyst reuse
After the first run at 143 °C, the solids were filtered and directly added to a second run at the same temperature. In this case the conversion was 57%. Isolation of the catalyst after this run, followed by washing with aqueous methanol, methanol and drying at 90 °C reactivated the catalyst, and a subsequent run yielded 86% of product (Table 1). This regeneration process could be repeated five times, without appreciable loss of activity. Thus, catalyst reuse is possible after isolation and washing of the catalyst. This washing step presumably is effective in removing the waste inorganics from the catalyst which begin to block access to the active sites.Extension to other substrates
The optimal reaction conditions were applied to a series of substituted halobenzenes and the results are tabulated in Table 2.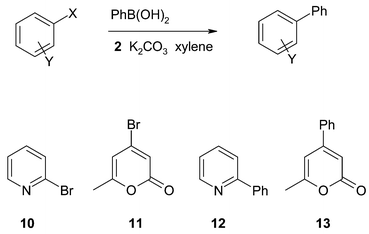
| compound | X | Y | Conditions | Yield |
|---|---|---|---|---|
| 3 | Br | 4-Me | 143 °C, 1 h | 89% |
| 4 | Br | 4-MeO | 143 °C, 1 h | 92% |
| 5 | Br | 4-CN | 143 °C, 1 h (3 h) | 68% (78%) |
| 6 | Br | 2-NO2 | 143 °C, 1 h (6 h) | 35% (36%) |
| 7 | Br | 4-NO2 | 143 °C, 1 h (6 h) | 3% (3%) |
| 8 | I | H | 143 °C, 1 h | 85% |
| 9 | Cl | H | 143 °C, 1 h (18 h) | 2% (3%) |
| 10 | — | — | 143 °C, 6 h | 74% |
| 11 | — | — | 143 °C, 4 h | 81% |
As can be seen, good to excellent yields are obtained for a range of aromatic substrates, with slightly better results being obtained for substrates with electron donating substituents, although nitro-containing substrates caused problems and appeared to decompose the catalyst leaving a black solid behind. In all other cases high yields were achieved, with excellent selectivity, generally in short reaction times. Bromo and iodo compounds both are active but, as expected, chlorobenzene was virtually inert.
Comparative reactions have been carried out with a homogeneous equivalent of the supported catalyst (prepared from butylamine, pyridine-2-aldehyde and Pd(OAc)2).17 While the initial rate of reaction was slightly more rapid than the supported version, the catalyst deactivated quickly (TON = 75 for PhBr + PhB(OH)2), and the separation of product and catalyst was difficult. While a detailed comparison of the homogeneous and heterogeneous catalysts has not been attempted, the similar rates of reaction indicate that diffusion is not a major problem for 2 under the conditions used here, and the extended lifetime may be a feature of the Pd being tethered to the support at a low density, stopping deactivation through clustering. Thus the supported versions of the catalysts do indeed allow for a more efficient reaction and for simpler and cleaner separation of product and recovery/reuse of catalyst.
2-Bromopyridine 10 was also investigated as a representative of a class of substrates which have found much use in the synthesis of multidentate ligands18–20 and in pharmaceutical applications.21,22 Our system gave a 74% yield of 2-phenylpyridine 12 from the Suzuki coupling after 6 hours, indicating that it is a good catalyst for such systems. Literature yields18–22 for this type of substrate vary considerably from poor to very good.
Bromopyrones are currently of interest in the synthesis of 4-substituted pyrones, which are showing promise as anti-cancer and anti-microbial agents.23 Suzuki and Sonagashira coupling methodologies have previously been applied successfully to these syntheses using homogeneous Pd catalysts. Using our chitosan-supported catalyst, the bromopyrone 11 was converted to the 4-phenyl derivative 13 in high yield after chromatographic separation. Around 10% of the unchanged 4-bromopyrone was recovered after chromatography.
Reactions at lower temperature
Since the analogous pyridylimine-Pd silica-based catalysts demonstrate good activity even at 90 °C, it was decided to investigate the reasons for the lower conversions achieved with 2 at temperatures below 143 °C. It was clear that as the temperature was reduced, the conversions dropped too, and that no further conversion occurred after ca. 1 h. A mass balance on bromobenzene indicated that no side reactions were occurring with this reaction partner, but it was found, by 11B CP MAS NMR of the entire reaction mixture after solvent removal, that the only boron-containing species present was borate. This was true for all temperatures, and indicated that extensive destructive decomposition of benzene boronic acid was occurring. Addition of extra boronic acid portionwise over several hours led to further product formation, consistent with the NMR results. Further addition of other reaction components (base, catalyst, bromobenzene), on the other hand, had no effect.Whilst the majority of papers on the Suzuki reaction do not mention this side reaction at all, a few do discuss such reactions, although in little detail. Protodeborylation of aryl boronic acids is known to occur, typically under acidic conditions (either aqueous acids or carboxylic acids under non-aqueous conditions.24–27) However, deborylation of arylboronic acids has occasionally been reported under basic conditions, for example during Suzuki coupling in the synthesis of a natural product.28 In this case, aqueous K2CO3 was used, indicating that protodeborylation can occur (in this case to ca. 40%) under basic conditions. These conditions often lead to excellent yields, although, as in almost all examples of Suzuki reactions, the boronic acid is used in excess. Other comments in the literature indicate that, while protodeborylation is often suppressed by using anhydrous conditions,29,30 in some cases it can occur to similar extents with or without water.31 Given that the more expensive and less readily synthesised boronic acid is the reagent used in (10–50%) excess, one can surmise that the majority of Suzuki reactions proceed with some protodeborylation. This side reaction therefore appears to be a significant difficulty in the development of truly green Suzuki methodology.
A particularly curious feature of our system is that protodeborylation is prevalent at low temperatures, but decreases significantly in importance as the temperature increases (no unreacted boronic acid can be found in any of the reactions after 6 h). Nothing in the literature could be found to indicate such a dramatic swing in selectivity with temperature. Work is in progress aimed at casting more light on this curious effect.
The Heck reaction
The Heck reaction was also investigated at substrate to Pd molar ratios of 1325 and 650. Under the first of these conditions, iodobenzene and butyl acrylate gave an 82% yield of butyl cinnamate in 42 h at 100 °C. With the higher catalyst quantity, an 87% yield was obtained after 20 h. Similar activity was also found with the same catalyst system immobilised on silica.32 Coupling of styrene and bromobenzene was also successfully accomplished at a substrate ∶ Pd ratio of 1420, giving an 88% yield of stilbene after 42 h, with a TON of 1250. Doubling the amount of catalyst led to a similar yield (85%) in 20 h.Conclusions
Chitosan-based Pd catalysts have been successfully prepared in a stable form. These catalysts prove to be active and reusable catalysts for both the Suzuki and Heck reactions. High yields of aryl-coupled products can be obtained in short times – these include model reactions on bromopyrones which are relevant to anti-cancer therapies. An unusual twist in the selectivity profile means that protodeborylation appears to be the dominant pathway at lower temperatures, but is almost absent at higher temperatures. Efficient recovery and reuse has been demonstrated.This work, along with other published work, indicates that chitosan is a particularly interesting support for catalysis – its stability, lower acidity (compared to supports such as silica), its film-forming properties, which can be exploited in intensive processing reactors, and its inherent chirality will form the basis for further applications of this fascinating material.
Acknowledgements
D. J. M. thanks the Royal Society for a Fellowship, S. H. thanks the EU for funding under the Erasmus Scheme. The authors are grateful to Dr I. J. S. Fairlamb for a gift of bromopyrone and useful discussionsReferences
- Handbook of Green Chemistry and Technology ed. J. H. Clark and D. J. Macquarrie, Blackwell, Oxford, 2002 Search PubMed.
- P. T. Anastas, M. M. Kirchhoff and T. C. Williamson, Appl. Catal., A: Gen., 2001, 221, 3 CrossRef CAS.
- M. N. V. Ravi Kumar, React. Funct. Polym., 2000, 46, 1 CrossRef.
- T. Vincent and E. Guibal, Ind. Eng, Res, Chem., 2002, 41, 5158 Search PubMed.
- L. M. Tang, M. Y. Huang and Y. Y. Jiang, Chin. J. Polym. Sci., 1996, 14, 57 Search PubMed.
- Y. An, D. Yuan, M. Y. Huang and Y. Y. Jiang, Macromol. Symp., 1994, 80, 257 CAS.
- L. M. Tang, M. Y. Huang and Y. Y. Jiang, Macromol. Rapid Commun., 1994, 15, 527 CrossRef CAS.
- H. S. Han, S. N. Jiang, M. Huang and Y. Y. Jiang, Polym. Adv. Technol., 1996, 7, 704 CrossRef CAS.
- M.-Y. Yin, G.-L. Yuan, Y.-Q. Wu, M.-Y. Huang and Y.-Y. Liang, J. Mol. Catal. A, 1999, 147, 93 CrossRef CAS.
- F. Quignard, A. Choplin and A. Domard, Langmuir, 2000, 16, 9106 CrossRef CAS.
- P. Buisson and F. Quignard, Aust. J. Chem., 2002, 55, 73 CrossRef CAS.
- W. Sun, C.-G. Xia and H.-W. Wong, New J. Chem., 2002, 26, 755 RSC.
- Y. Chang, Y. P. Wang and Z. X. Su, J. Appl. Polym. Sci., 2002, 83, 2188 CrossRef CAS.
- D. Hu, Y. Cui, X. Dong and Y. Fang, React. Funct. Polym., 2001, 48, 201 CrossRef CAS.
- E. B. Mubofu, J. H. Clark and D. J. Macquarrie, Green Chem., 2001, 3, 23 RSC.
- R. B. Bedford, C. S. J. Cazin, M. B. Hursthouse, M. E. Light, K. J. Pike and S. Wimperis, J. Organomet. Chem., 2001, 633, 173 CrossRef CAS.
- E. B. Mubofu, D.Phil Thesis, University of York, 2001.
- F. Lam, M. Q. Feng and K. S. Chan, Tetrahedron, 1999, 55, 8377 CrossRef CAS.
- J. K. Judice, S. J. Keipert and D. J. Cram, J. Chem. Soc., Chem. Commun., 1993, 1323 RSC.
- A. Puglisi, M. Benaglia and G. Roncan, Eur. J. Org. Chem., 2003, 8, 1552 CrossRef.
- M. Nettekoven, Synlett, 2001, 1917 CrossRef CAS.
- P. Meier, S. Legraverant, S. Muller and J. Schaub, Synthesis, 2003, 551 CrossRef CAS.
- L. R. Marrison, J. M. Dickinson and I. J. S. Fairlamb, Bioorg. Med. Chem. Lett., 2002, 12, 3509 CrossRef CAS.
- D. S. Matteson, The Chemistry of the Metal–Carbon Bond ed. F. R. Hartley, Wiley, Chichester, 1987, ch. 3, pp. 307 Search PubMed.
- H. Kuivala and K. V. Nahabadian, J. Am. Chem. Soc., 1961, 83, 2159 CrossRef CAS.
- H. Kuivala and K. V. Nahabadian, J. Am. Chem. Soc., 1961, 83, 2164 CrossRef CAS.
- H. Kuivala and K. V. Nahabadian, J. Am. Chem. Soc., 1961, 83, 2167 CrossRef.
- D. Muller and J.-P. Fleury, Tetrahedron Lett., 1991, 32, 2229 CrossRef CAS.
- A. B. Charette and A. Giroux, J. Org. Chem., 1996, 61, 8718 CrossRef.
- E. J.-G. Anctil and V. C. Snieckus, J. Organomet. Chem., 2002, 653, 150 CrossRef CAS.
- I. C. Christoforou, P. A. Koutentis and C. W. Rees, Org. Biomol. Chem., 2003, 1, 2900 RSC.
- E. B. Mubofu, J. H. Clark and D. J. Macquarrie, Green Chem., 2000, 2, 53 RSC.
| This journal is © The Royal Society of Chemistry 2004 |