Rare-earth metal triflates as versatile catalysts for the chloromethylation of aromatic hydrocarbons
Toru Kishidaa, Takayoshi Yamauchia, Yoshihiro Kubotab and Yoshihiro Sugi*b
aK. K. Nissei Kagaku Kogyosho, 2-18-110 Juhachijyo, Yodogawa-ku, Osaka 532-0001, Japan
bDepartment of Materials Science and Technology, Faculty of Engineering, Gifu University, Gifu 501-1193, Japan. E-mail: sugi@apchem.gifu-u.ac.jp
First published on 8th December 2003
Abstract
Rare-earth metal triflates, such as Sc(OTf)3, Yb(OTf)3, and Sm(OTf)3 work as highly effective catalysts for the chloromethylation of aromatic hydrocarbons with hydrochloric acid and trioxane. They are active enough in aqueous solution at a concentration of less than 1–5% of the substrate under heterogeneous conditions of organic and aqueous phases. The triflate stays in the aqueous phase after the catalysis, and the organic products are easily separated from the catalyst. Sc(OTf)3 was stable under the reaction conditions. The catalyst in the aqueous solution could be recycled and used for further reactions without significant loss of activity. The chloromethylation of m-xylene catalyzed by Sc(OTf)3 gave 2,5-bis(chloromethyl)-m-xylene with 1,2,4,5-substitution, which is expected to be a key intermediate for pyromellitic dianhydride. The catalysis occurred via the formation of a chloromethylated triflate complex, and electrophilic addition to an aromatic hydrocarbon.
Green ContextThe chloromethylation of aromatic molecules is a very useful reaction since the products can be used as intermediates on route to many valuable products. The reaction of the aromatic substrate with a formaldehyde source such as trioxane and hydrochloric acid is a popular and atom economic route but catalysts are required to achieve reasonable reaction rates and traditionally these have been required in large, often stoichiometric quantities and cannot easily be recovered. Here it is shown that metal triflates are effective catalysts for aromatic chloromethylations under atom-efficient conditions. The catalysts are active at very low concentrations and are easily recoverable.JHC |
1 Introduction
Chloromethyl substituted aromatic hydrocarbons are promising key intermediates because of easy transformation to many chemicals such as fine-chemicals, pharmaceuticals, and polymers. The chloromethylation of aromatic hydrocarbons has been well documented in previous papers.1–6 The reaction of aromatic hydrocarbons with hydrochloric acid and trioxane or paraformaldehyde as a formaldehyde precursor sometimes gave the chloromethylated products without a catalyst,5,6 however, the rate is slow and insufficient for practical chemical processes. Lewis acids such as zinc chloride, stannic chloride, and boron trifluoride are well known catalysts for the reaction; among these acids, zinc chloride is an effective catalyst in hydrochloric acid solution,1,2 however, a stoichiometric amount of catalyst to substrate is required, making the work up procedure tedious. These catalysts, in general, suffer from the inherent problems of corrosiveness, high susceptibility to water, difficulty in catalyst recovery, environmental hazards, waste control after the reaction, etc. It is important to replace these highly corrosive, hazardous and polluting acid catalysts with environmentally conscious catalysts which are active under mild conditions, and can be easily recovered after the reactions and reused for new reactions.7 Recently, it has been shown that rare-earth metal triflates act as Lewis acids in aqueous medium; they are stable in water, and are active catalysts for organic synthesis.8–12 In this paper, we report that some rare-earth metal triflates are highly active for the chloromethylation of many aromatic hydrocarbons.2 Experimental
2.1 Reagents
Scandium, ytterbium, samarium, hafnium, and indium triflates were purchased from Tokyo Kasei Chem. Ind., Co. Ltd., Tokyo, Japan. Zinc chloride and trioxane were obtained from Kishida Chemicals Co. Ltd., Osaka, Japan.2.2 Reaction procedures
Typical procedures for the chloromethylation are shown in the case of m-xylene as follows: a mixture of m-xylene (I; 1.0 g, 9.4 mmol), trioxane (1.3 g, 14.1 mmol), 35% aqueous hydrochloric acid (4.9 g, 47 mmol), and scandium triflate (50 mg, 0.094 mmol) was stirred in a 30 ml round flask for 5 h at 70 °C. After cooling, the organic products were extracted with cyclohexane, and dried over anhydrous Na2SO4. The solvent was evaporated in vacuo, and the organic residue was resolved in cyclohexane again, and analyzed by hlpc using Finepack SIL C-18-5 φ 4.6 × 250 mm (JASCO, Tokyo, Japan). The yields of chloromethyl-2,4-dimethylbenzene (II) and 1,5-bis(chloromethyl)-2,4-dimethylbenzene (III) were 73 and 20%, respectively. Each product was separated by silica gel column chromatography, and identified by 1H and 13C NMR, IR, and elemental analysis. Residual aqueous catalytic solution was evaporated in vacuo to a white solid, which was dissolved in a small amount of water, and treated with activated carbon. The filtrate was evaporated to dryness in vacuo to give scandium triflate in 94% yield. Scandium triflate was identified by IR in comparison with an authentic sample.The products from o-xylene and pseudocumene were identified by the comparison of NMR spectra of original and hydrogenated products. Properties of 1,2,4,5-substituted products from o-, m-, and p-xylene isomers are as follows:
The amount and efficiency of HCl and CH2O in the organic product were calculated on the basis of the comparison with introduced amounts.
2.3 Recycling of catalyst
The reaction catalyzed by Sc(OTf)3 was carried out in the same manner as in the previous section. After the separation of the organic layer, the aqueous catalytic solution was saturated with HCl by bubbling with dry HCl gas, and subjected to further reaction. The reaction was carried out by the addition of m-xylene (1.0 g) and trioxane (1.3 g) for 5 h at 70 °C. The analysis of products was carried out as described previously.3 Results and discussion
The chloromethylation of m-xylene with hydrochloric acid and trioxane as a formaldehyde precursor was examined, using a rare-earth metal triflate as a catalyst. Typical results are shown in Fig. 1. m-Xylene gave chloromethyl-2,4-dimethylbenzene (II) and 1,5-bis(chloromethyl)-2,4-dimethylbenzene (III) without a catalyst, however, the combined yield of II and III was as low as 20%. On the other hand, rare-earth triflates such as scandium, ytterbium, samarium, indium, and hafnium triflates are excellent catalysts for the chloromethylation of m-xylene with hydrochloric acid and trioxane. Triflates are sufficiently active for the chloromethylation of m-xylene in catalytic amounts. The conversion of m-xylene is in the order: Sc(OTf)3 ≈ Yb(OTf)3 ≈ Sm(OTf)3 ≫ Hf(OTf)3 > In(OTf)4 > none; however, the yield of III decreased in the order: Sc(OTf)3 > Yb(OTf)3 > Sm(OTf)3 ≫ Hf(OTf)3 > In(OTf)4 > none. From these results, Sc(OTf)3, Yb(OTf)3, and Sm(OTf)3 have higher activity among the triflates for the chloromethylation. The catalysis occurred under heterogeneous conditions of organic and aqueous phases: the catalysts were easily separated from the reaction mixtures after the reaction, and recycled for new reaction without significant loss of activity. The chloromethylation using ZnCl2 as catalyst gave the products at the same level; however, it was necessary to use an approximately stoichiometric amount for corresponding yields. Hereafter, we chose Sc(OTf)3 as the catalyst in experiments to elucidate the factors controlling the catalysis, because it possessed the highest activity among the triflates. | ||
| Fig. 1 Chloromethylation of m-xylene catalyzed by rare-earth metal triflates. Reaction conditions: rare-earth metal triflate, 0.94 mmol; m-xylene, 1.0 g (9.42 mmol); conc. HCl, 4.9 g (47 mmol); trioxane 0.4 g (4.7 mmol); temperature, 70 °C; period, 5 h. | ||
The effects of reaction temperature on the chloromethylation are shown in Fig. 2 (trioxane/m-xylene = 0.5). The catalytic activity increased with temperature. The reaction was enhanced significantly around 50–60 °C. The yield of II increased with temperature, reached a maximum at 70 °C, and then decreased above 80–90 °C. However, the yield of III increased with temperature. The formation of tris(chloromethyl)-2,4-dimethylbenzene increased as the yield of II decreased at higher temperatures.
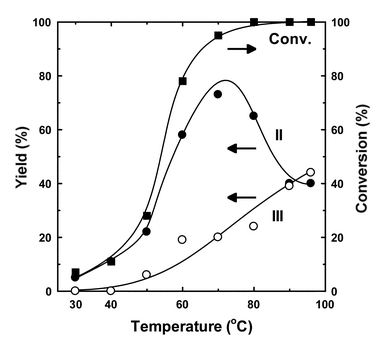 | ||
| Fig. 2 Effects of reaction temperature on the chloromethylation of m-xylene catalyzed by Sc(OTf)3. Reaction conditions: Sc(OTf)3, 0.05 g (0.94 mmol); m-xylene, 1.0 g (9.4 mmol); conc. HCl, 4.9 g (47 mmol); trioxane 0.4 g (4.7 mmol); period, 5 h. | ||
The effect of the concentration of hydrochloric acid at 70 °C is shown in Fig. 3a. No reaction occurs in the absence of hydrochloric acid. The activity was a function of the concentration of hydrochloric acid: an increase of the concentration increased the formation of II and III. The rate was saturated at an HCl concentration of around 20% (HCl/m-xylene = ∼ 3). However, the yield of III increased slightly with the decrease of that of II at higher HCl concentration. These results show that the concentration of hydrochloric acid solution is not the principal factor controlling the catalysis. Fig. 3b shows the amount of HCl and formaldehyde in the products, and their efficiencies during the reaction. An increase of the amount of HCl enhanced the amount of CH2O and HCl in the products. However, the efficiency of CH2O increased with the concentration of hydrochloric acid although the efficiency of HCl for the reaction decreased. These results show that the activation of formaldehyde or its hydrate is not involved in the acid catalysis of triflate, and that HCl plays an important role in the reaction: the formation of chloromethanol (ClCH2OH) and subsequent formation of chloromethyl complex are key steps for the chloromethylation.
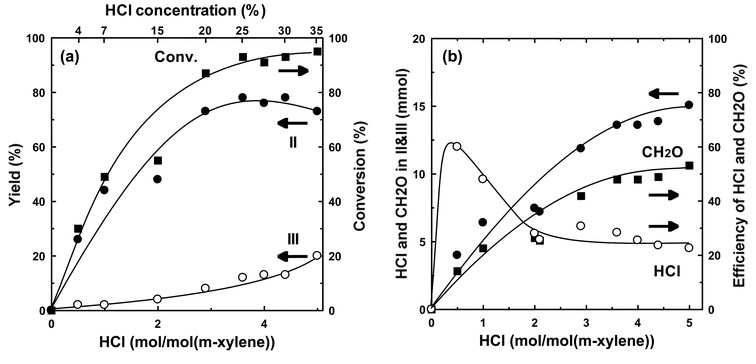 | ||
| Fig. 3 Effects of the concentration of hydrochloric acid on the chloromethylation of m-xylene catalyzed by Sc(OTf)3. (a) catalytic feature. (b) balance of CH2O and HCl. Reaction conditions: Sc(OTf)3, 0.05 g (0.094 mmol); m-xylene, 1.0 g (9.4 mmol); conc. HCl, 4.9 g (47 mmol); trioxane 0.4 g (4.7 mmol); temperature, 70 °C; period, 5 h. | ||
Fig. 4a shows the effects of the ratio of trioxane and m-xylene. The conversion of m-xylene was high enough for the formation of II even with the ratio of trioxane/m-xylene = 0.5 (formaldehyde/m-xylene = 1.5), and the yield of II was around 70%. The addition of trioxane in excess amount enhanced the formation of III, and reached the maximum yield of ca. 70% at a trioxane/m-xylene ratio of 4. Further addition of trioxane decreased the formation of III to yield polychloromethylated products as a by-product. Fig. 4b shows that the amount of formaldehyde and hydrochloric acid in the products was maximized at a ratio of trioxane/m-xylene = 1.5 (formaldehyde/m-xylene = 4.5), and decreased with further addition of trioxane. However, the efficiency of CH2O was decreased with the increase of its concentration, whereas the efficiency of HCl remained almost constant with the increasing amount of CH2O. It is considered that trioxane forms formaldehyde before the reaction with m-xylene. These results indicate that the activation of formaldehyde is a key step for enhancement of the chloromethylation.
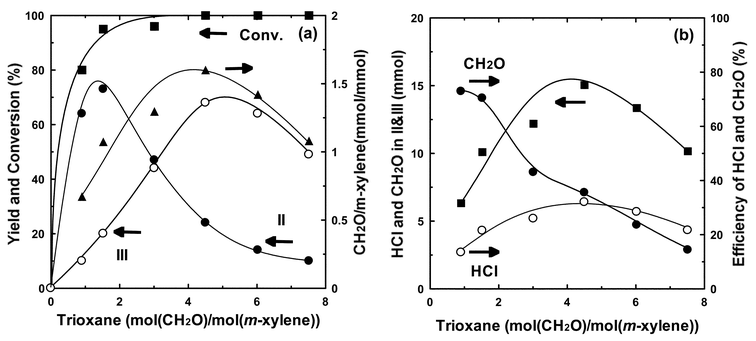 | ||
| Fig. 4 Effects of the ratio of trioxane to m-xylene on the chloromethylation of m-xylene catalyzed by Sc(OTf)3. (a) catalytic feature. (b) balance of CH2O and HCl. Reaction conditions: Sc(OTf)3; 0.05 g (0.094 mmol); m-xylene, 1.0 g (9.4 mmol); conc. HCl, 4.9 g (47 mmol); temperature, 70 °C ; period, 5 h. | ||
Fig. 5 shows that the amount of the catalyst is not a crucial issue for the enhancement of the catalysis, although the rate was rapidly increased up to 1 mol% of the catalyst.
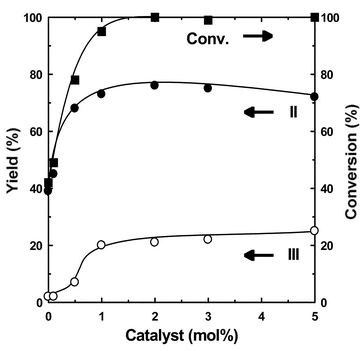 | ||
| Fig. 5 Effects of catalyst amount on the chloromethylation of m-xylene catalyzed by Sc(OTf)3. Reaction conditions: Sc(OTf)3, 0.0–0.25 g (0.094–0.47 mmol); m-xylene, 1.0 g (9.4 mmol); conc. HCl, 4.9 g (47 mmol); trioxane 0.4 g (4.7 mmol); temperature, 70 °C; period, 5 h. | ||
The catalysis under our conditions occurs in a heterogeneous mixture composed of m-xylene, trioxane, and triflate in aqueous hydrochloric acid. The organic compounds were added to accelerate the reaction by improvement of the contact of the reactant with the triflate in the aqueous phase. Fig. 6 shows the effect of some organic additives. Formic and acetic acids enhanced the formation of III. However, the addition of nitrobenzene and methylcyclohexane decreased the activity, and the yield of III was decreased. The addition of dioxane also promoted the formation of III. The amount of acetic acid was examined to elucidate the effect of acid addition. Fig. 7 shows the effects of acetic acid on the chloromethylation. A small amount of acetic acid enhanced the yield of III; however, further addition decreased the yield of III. These results show that acetic acid promotes the catalysis effectively by addition in a small amount; however, it retards the catalysis by the further addition.
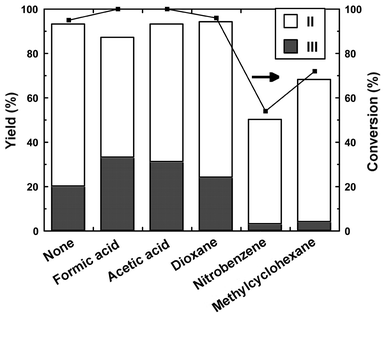 | ||
| Fig. 6 Effects of organic additives on the chloromethylation of m-xylene catalyzed by Sc(OTf)3. Reaction conditions: Sc(OTf)3, 0.05 g (0.094 mmol); m-xylene, 1.0 g (9.4 mmol); conc. HCl, 4.9 g (47 mmol); trioxane 0.4 g (4.7 mmol); additive, 1.0 ml; temperature, 70 °C; period, 5 h. | ||
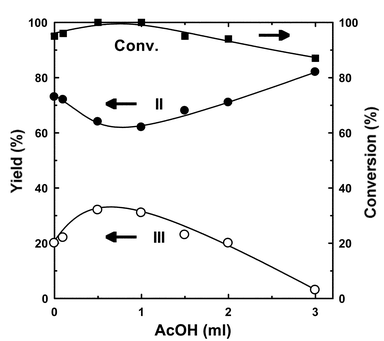 | ||
| Fig. 7 Effects of acetic acid on the chloromethylation of m-xylene catalyzed by Sc(OTf)3. Reaction conditions: Sc(OTf)3, 0.05 g (0.094 mmol); m-xylene, 1.0 g (9.4 mmol); conc. HCl, 4.9 g (47 mmol); trioxane 0.4 g (4.7 mmol); temperature, 70 °C; period, 5 h. | ||
It is important to evaluate the recyclability of catalysts. We examined the recovery and recyclability of Sc(OTf)3 catalyst using two methods. First, the catalyst was recycled and reused as aqueous solution for further reaction. After the reaction was finished, the aqueous catalytic solution was recovered by separation of the organic layer. The aqueous solution was saturated with HCl by bubbling with HCl gas, and the reaction was carried out using recovered catalytic solution by the addition of m-xylene and trioxane. The change in catalyst performance is summarized in Table 1. The catalytic activity decreased gradually by recycling to result in a decrease of the yield of II and III. The decrease should be due to incomplete recovery of the catalyst, and also to the different reaction conditions from the original process. Optimization of conditions will improve the yield of recycled reactions. Secondly, recovery of Sc(OTf)3 was examined by evaporation of the catalytic solution. The residual solid was treated with activated charcoal to give Sc(OTf)3 in high yield. Recovered triflate can be used as fresh catalyst for the chloromethylation. These results show that the triflate is not hydrolyzed in the presence of an excess of hydrochloric acid. These two methods of catalyst recovery indicate that triflate is stable under the reaction conditions and easily recycled after the reactions.
| Catalytic cycle | Conversion (%) | Yield (%) | |
|---|---|---|---|
| II | III | ||
| Reaction conditions: m-xylene, 1.0 g (9.4 mmol); trioxane 0.43 g (4.7 mmol); Sc(OTf)3, 0.046 g (0.094 mmol); temperature, 70 °C; period, 5 h. | |||
| 1st | 95 | 73 | 20 |
| 2nd | 79 | 65 | 11 |
| 3rd | 70 | 62 | 7 |
Table 2 summarizes the application of Sc(OTf)3 to the chloromethylation of aromatic hydrocarbons. Although the reaction was carried out under relatively mild conditions such as 70 °C, and at a molar ratio of trioxane/alkylbenzene of 0.5 (formaldehyde/alkylbenzene = 1.5), benzene, toluene, xylenes, mesitylene, pseudocumene, and durene gave the chloromethylated product in good to excellent yields. The hydrocarbons gave monochloromethylated hydrocarbons in high yield, and also gave disubsutituted products in low yield. The product distributions are similar to the conventional zinc chloride method: the substitution occurred in the o- and p-direction. The less bulky isomer was predominant, especially for the monochloromethylation. The difference in the products for the xylene isomers is interesting. m-Xylene gave III selectively, whereas p-xylene also gave 1,4-bis(chloromethyl)-2,5-dimethylbenzene. The introduction of two chloromethyl groups to the m- and p-xylenes occurred selectively at the neighbor of the methyl groups to yield the dichloromethylated products with 1,2,4,5-substitution. On the other hand, the dichloromethylated products from o-xylene are 1,2-bis(chloromethyl)-4,5-dimethylbenzene and 1,4-bis(chloromethyl)-2,3-dimethylbenzene. The chloromethylation of pseudocumene also gave two isomers, 1-chloromethyl-2,4,5-trimethylbenzene and 1-methyl-2,3,6-trimethylbenzene, and the formation of the product with 1,2,4,5-substituted products was not selective. Cumene and t-butylbenzene showed low activity for the reaction. Bromobenzene and benzonitrile were also inactive for the chloromethylation.
| Entry | Substrate | Conv. (%) | Products and yield (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Reaction conditions: substrate, 1 g; (HCHO)3, 0.5 equiv.; conc. HCl, 5 equiv.; Sc(OTf)3, 1 mol%; temperature, 70 °C; period, 5 h. | ||||||||||
| 1 |  | 73 | 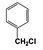 | 67 | 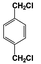 | 3 | — | — | — | — |
| 2 | 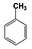 | 100 | 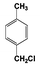 | 37 | 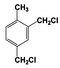 | 32 | — | — | — | — |
| 3 | 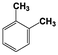 | 100 | 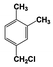 | 35 | 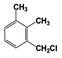 | 11 | 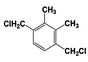 | 28 | 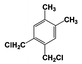 | 22 |
| 4 | 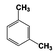 | 95 |  | 73 | 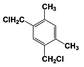 | 20 | — | — | — | — |
| 5 | 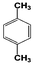 | 92 | 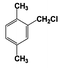 | 69 | 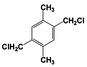 | 19 | — | — | — | — |
| 6 | 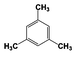 | 100 | 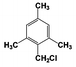 | 70 | 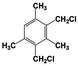 | 30 | — | — | — | — |
| 7 | 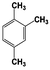 | 100 | 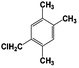 | 48 | 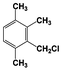 | 10 |  | 33 | — | — |
| 8 | 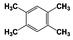 | 91 | 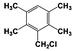 | 74 | 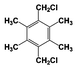 | 16 | — | — | — | — |
| 9 | 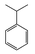 | 26 | 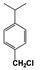 | 17 | 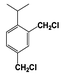 | 4 | — | — | — | — |
| 10 | 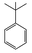 | 9 | 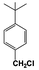 | 6 | — | — | — | — | — | — |
| 11 | 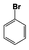 | 0 | — | — | — | — | — | — | — | — |
| 12 |  | 0 | — | — | — | — | — | — | — | — |
Scheme 1 shows a plausible mechanism for the catalysis. First, trioxane yields formaldehyde by acid catalysis of hydrochloric acid. Then, formaldehyde and HCl react with triflate to yield chloromethylated triflate complex (X). The formation of the complex should occur either way:
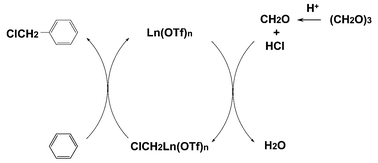 | ||
| Scheme 1 Plausible mechanism. | ||
(A) Formaldehyde and HCl give the chloromethanol and react with triflate to yield X.
(B) The hydroxymethyl triflate complex is formed from formaldehyde, and then OH is substituted with Cl to form X.
Mechanism (A) is more plausible than (B) because no reaction occurs in the absence of hydrochloric acid. Complex X reacts with aromatic hydrocarbon to yield the chloromethylated product and triflate by electrophilic substitution. This mechanism is close to the classical zinc chloride catalysis.1
4 Conclusions
Rare-earth metal triflates are highly active for the chloromethylation of various aromatic hydrocarbons. The most important characteristic is that a catalytic amount of triflate was effective for the chloromethylation. Two or three times the amount of hydrogen chloride compared to chloromethyl groups in the products is sufficient for reasonable conversion of hydrocarbons. The catalysis by triflates easily occurs in a heterogeneous mixture of hydrocarbon, trioxane, and triflate in aqueous hydrochloric acid. Since the triflate stays in the aqueous phase during the reaction, the catalyst was easily separated from the products, and recycled for further reaction. The catalyst was stable under the reaction conditions.From an environmental point of view, rare-earth metal triflates are active enough for the green chloromethylation of aromatic hydrocarbons. Further aspects of the catalysis and the application to organic syntheses and practical chemical processes are under investigation.
References
- R. C. Fuson and C. H. McKeever, in Organic Reactions, John Wiley, New York, 1943, vol. 1, pp. 63–90 Search PubMed.
- G. A. Olah, in Friedel Crafts and Related Reactions, John Wiley, New York, 1964, vol. 2, part 2, pp. 659–784 Search PubMed.
- J. H. Wood, M. A. Perry and C. C. Tung, J. Am. Chem. Soc., 1950, 72, 2989 CrossRef CAS; J. H. Wood, M. A. Perry and C. C. Tung, J. Am. Chem. Soc., 1950, 72, 2992 CrossRef CAS.
- C. D. Shacklett and H. A. Smith, J. Am. Chem. Soc., 1951, 73, 766 CrossRef CAS.
- T. Sasaki, Y. Hasebe, S. Okada and T. Arai, Jpn. Kokai Tokkyo Koho JP63-048234, 1988.
- M. Gerisch, J. R. Krumper, R. G. Bergman and T. Don Tilley, Organometallics, 2003, 22, 47 CrossRef CAS.
- Green Chemistry. Frontiers in Benign Chemical Synthesis and Processes, ed. P. T. Anastas and T. C. Williamson, Oxford University Press, Oxford, 1998 Search PubMed.
- S. Kobayashi, Synlett, 1994, 689 CrossRef CAS.
- S. Kobayashi, in Green Chemistry, ed. M. Misono and S. Murahashi, Kodansha Scientific, Tokyo, 2002, ch. 12, pp. 164–176 Search PubMed.
- A. G. M. Barett and D. C. Braddock, Chem. Commun., 1997, 351 RSC.
- F. J. Waller, A. G. Barett, D. C. Braffock and D. Ramprasad, Chem. Commun., 1997, 613 RSC.
- F. J. Waller, A. G. Barett, D. C. Braffock and D. Ramprasad, Tetrahedron Lett., 1998, 39, 1641 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2004 |