Marked enantioselectivity enhancements for Diels–Alder reactions in ionic liquids catalysed by platinum diphosphine complexes
Simon Doherty*a, Peter Goodrichb, Christopher Hardacre*ab, He-Kuan Luoa, David W. Rooneybc, Kenneth R. Seddonab and Peter Styringd
aSchool of Chemistry, Queen's University Belfast, Belfast, Northern Ireland, UK BT9 5AG. E-mail: c.hardacre@qub.ac.uk, s.doherty@qub.ac.uk
bQUILL, Queen's University Belfast, Belfast, Northern Ireland, UK BT9 5AG
cSchool of Chemical Engineering, Queen's University Belfast, Belfast, Northern Ireland, UK BT9 5AG
dDepartment of Chemical and Process Engineering, University of Sheffield, Sheffield, UK S1 3AD
First published on 8th December 2003
Abstract
Asymmetric Diels–Alder reactions using platinum complexes of BINAP, or of conformationally flexible NUPHOS-type diphosphines, have been compared in dichloromethane and selected ionic liquids. Significant enhancements in the enantioselectivity (Δee ≈ 20%), as well as reaction rate, were achieved in ionic liquids compared with the organic media.
Green ContextThere has been an extraordinary growth in the research work on ionic liquids in recent years. These fascinating substances offer the potential for carrying out many chemical processes without the need for environmentally threatening volatile organic solvents. Here their use as a VOC substitute is made more useful through an improvement in reaction selectivity. Diels–Alder reactions in ionic liquids have previously been demonstrated but here useful improvements in enantioselectivity are also achieved.JHC |
Introduction
In general, the design of asymmetric catalysts has been based upon conformationally-restricted, enantiopure ligands which impart their chirality on a transformation to achieve high enantioselectivities. Recently, this concept has been challenged by the discovery of asymmetric activation, a process that involves activation of an enantiopure catalyst with a chiral activator, selective activation of one enantiomer of a racemic catalyst, and more recently the formation of highly effective catalysts based on a conformationally-flexible ligand coupled with a chiral activator.1 For example, Mikami and Matsukawa have used BINOLate-titanium based catalysts activated with enantiopure 1,1′-bi-2-naphthol (BINOL) to achieve an ee of 90% in the asymmetric carbonyl-ene reaction,2 and 84% in the asymmetric Diels–Alder reaction of the Danishefsky diene with glyoxylate.3 Becker et al. have also shown that coordination of 1,1′-bis(diphenylphosphino)biphenyl (BIPHEP) to platinum significantly slows atropinversion such that the enantiopure Lewis acid fragment λ/δ-{Pt(BIPHEP)}2+, generated from either λ/δ-[(BIPHEP)Pt{(S)-BINOL}] or λ/δ-[(BIPHEP)PtCl2], is highly selective for the Diels–Alder reaction between acryloyl-N-oxazolidinone and cyclopentadiene, giving ee values as high as 94%.4Recently, Doherty and co-workers reported the synthesis of a new class of conformationally flexible diphosphine, R4-NUPHOS (Fig. 1), based on two diphenylphosphino- groups linked by a four-carbon conjugated tether.5 Preliminary studies have shown that the platinum group metal complexes of R4-NUPHOS diphosphines are highly active for Kumada cross-coupling reactions and the transfer hydrogenations of ketones.6 Recently, efficient Diels–Alder reactions between acryloyl-N-oxazolidinones and cyclopentadiene (Cp–H) catalysed by platinum and palladium complexes of NUPHOS-type diphosphines have been observed.7 Following the procedure reported by Becker et al.,4 enantiopure Lewis acids of the type δ-[(R4-NUPHOS)Pt(OTf)2] gave good conversions and high enantioselectivities for the low temperature Diels–Alder reaction between 1a or 1b and Cp–H, although notably the same catalysts gave low conversions and only moderate ee’s at 20 °C (Fig. 2). These low conversions appear to be due to rapid and quantitative formation of the catalytically inactive cation [(R4-NUPHOS)Pt(η5-Cp)]+ (vide infra). Becker et al. have noted that a similar η5-Cp complex also forms at low temperature;4 however, the Diels–Alder reaction between 1a and cyclopentadiene is fast compared with the formation of this cation, and therefore the reaction can proceed. In contrast, at room temperature, formation of the inactive complex dominates.
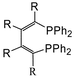 | ||
| Fig. 1 Schematic of R4-NUPHOS. | ||
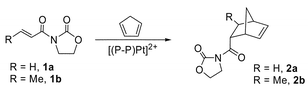 | ||
| Fig. 2 Asymmetric Diels–Alder reaction between acryloyl-N-oxazolidinone and cyclopentadiene. | ||
Solvent effects are known to have a strong influence on Diels–Alder reactions and, recently, ionic liquids have been reported to show large increases in reaction rates.8 Thus, we reasoned that ionic liquids could be used to overcome the problems found in dichloromethane by stabilising the catalyst with respect to deactivation and/or reducing the reactions times which would limit the extent of atropinversion.
Ionic liquids are one of a number of possible ‘green’ alternatives to conventional solvents due to their low vapour pressure. A wide range of reactions have been performed in ionic liquids to date including alkylations,9 C–C bond coupling reactions,10–12 polymerisations,13–15 hydrogenations,16–18 hydroformylation,16 and alkoxycarbonylation.19 These reactions have been extensively reviewed recently.20 Ionic liquids have also been used for asymmetric transformations; however, these have often been performed in the presence of a co-solvent, and in many cases without any beneficial effect on the enantioselectivity compared to conventional media. For example, Song and Roh demonstrated that Jacobsen's chiral [(salen)Mn(III)] ([N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediamine] manganese(III) chloride) epoxidation catalyst performed similarly in CH2Cl2 and a CH2Cl2-[bmim][PF6] mixture.21 Similar results have been reported for asymmetric ring opening of epoxides,22 asymmetric hydrogenations,23 asymmetric dihydroxylation of olefins24 and kinetic resolution of epoxides25 The ionic liquid does, however, allow the catalyst to recycle a number of times without reduction in the enantioselectivity. Jessop et al. have shown that ionic liquids can enhance the enantioselectivity in the asymmetric reduction of tiglic acid using ruthenium BINAP (2,2′-bis(diphenylphosphino)-1′1-binaphthyl) based catalysts.26 Using a range of ionic liquids, the ee’s reported were found to increase from 88% in MeOH to 95% in [emim][N(O2SCF3)2] ([emim][NTf2]). Baudequin et al have recently reviewed the effect of ionic liquids on enantioselective reactions.27
To date, only one asymmetric Diels–Alder reaction performed in ionic liquids has been reported. Meracz and Oh observed an ee of 96% for the Diels–Alder reaction of oxazolidinone 1b and Cp–H at room temperature using a rigid copper bisoxazoline-based chiral Lewis acid with a yield of 65% in 1,3-dibutylimidazolium tetrafluoroborate. This was compared with dichloromethane which showed only 76% ee with a yield of only 4%.28
In this paper we also report that ionic liquids provide an ideal medium in which to perform the room temperature Diels–Alder reaction between oxazolidinone 1a/b and Cp–H (Fig. 2) catalysed by platinum complexes of conformationally rigid (BINAP) and flexible (NUPHOS, BIPHEP) diphosphines. Mechanistic information is also presented which is used to understand the promoting effect of the ionic liquid.
Experimental
General procedures
1-Ethyl-3-methyimidazolium bis{(trifluoromethyl)sulfonyl}amide ([emim][NTf2]), 1-butyl-3-methylimidazolium bis{(trifluoromethyl)sulfonyl}amide ([bmim][NTf2]) and 1-methyl-3-octylimidazolium bis{(trifluoromethyl)sulfonyl}amide ([omim][NTf2]) were prepared following the method of Bonhôte et al.29 1-Butyl-3-methyl imidazolium hexafluorophosphate ([bmim][PF6]) was prepared following the method of Seddon and co-workers.30N-butyl-N-methylpyrollidinium bis{(trifluoromethyl)sulfonyl}amide ([C4mpyl][NTf2]) was prepared following the method of MacFarlane et al.31 The NUPHOS, BINAP and BIPHEP catalyst precursors (3, 5, 7 and 9) were prepared following methods reported by Doherty and co-workers,6a Gugger et al.32 and Tudor et al.,33 respectively. Oxazolidinones 1a and 1b were prepared using methods reported by Evans et al.34 Cyclopentadiene was distilled by cracking dicyclopentadiene and used immediately. 1H and 31P{1H} NMR spectra were recorded on Bruker AC 200, AMX 300 and DRX 500 spectrometers.All manipulations involving air-sensitive materials were carried out in an inert atmosphere glove box, or using standard Schlenk line techniques, under an atmosphere of dinitrogen or argon in oven-dried glassware. Diethyl ether and hexane were distilled from potassium/sodium alloy, tetrahydrofuran from potassium, dichloromethane from calcium hydride, and methanol from magnesium. Deuteriochloroform was pre-dried with calcium hydride, vacuum transferred, and stored over 4A molecular sieves. Unless otherwise stated, commercially purchased materials were used without further purification. Purification of reaction products was carried out by column chromatography on reagent silica gel (60–200 mesh). Analytical high performance liquid chromatography (HPLC) was performed on an Agilent 1100 series HPLC instrument, using HPLC grade hexane ∶ ethyl acetate ∶ isopropanol (94 ∶ 4 ∶ 2) as the eluent at 1 cm3 min−1 flow rate. Enantioselectivities were determined using a Diacel Chiralcel OD–H reverse phase column. The retention times of the endo enantiomers were 15.79 min (endo2S) and 17.21 min (endo2R). The absolute configuration of the endo cycloadduct was assigned by comparison with the retention times of samples prepared from (R)- and [{(S)-BINAP}PtCl2].35
Catalyst activation
Two methods of catalyst activation were used. In method (i), a solution of the platinum complex 3 or 5 (0.032 mmol, 20 mol%) and Ag[SbF6] (2.0 equiv.) in dichloromethane (2.0 cm3) was stirred under a dinitrogen atmosphere for 1 h at 20 °C, after which time the resulting solution was filtered using a filter cannula. In method (ii), a rapidly stirred solution of complex 7 or 9 (0.016 mmol, 10 mol%) in dichloromethane (ca. 2 cm3) was treated with HOTf (2.2 µL, 1.4 equiv.), resulting in an immediate colour change from deep yellow–orange to near colourless. The resulting mixture was stirred for 5 min at 20 °C.General procedure for enantioselective Diels–Alder reactions between cyclopentadiene and oxazolidinone in dichloromethane
A dichloromethane solution (2.0 cm3) of catalyst activated according to method (i) or (ii) was cooled to −78 °C and oxazolidinone 1a or 1b (0.16 mmol) and cyclopentadiene (1.2 mmol) added. The reaction mixture was allowed to warm to the desired temperature and left to stir. At the end of the reaction, the solution was quenched with (S,S)-1,2-diphenylethylenediamine (S,S-dpeda) and the dichloromethane removed under vacuum. The crude mixture was purified by column chromatography over silica gel (60–200 mesh, 30% ethyl acetate–hexane) and the products analysed by 1H NMR spectroscopy and HPLC. Conversions were calculated using samples taken directly from the reaction solution. All conversions reported are ±10%.General procedure for enantioselective Diels–Alder reactions between cyclopentadiene and oxazolidinone in ionic liquids
Ionic liquid (2.0 cm3) and oxazolidinone 1a or 1b (0.16 mmol) were added to a dichloromethane solution (2.0 cm3) of the catalyst prepared according to methods (i) or (ii). After stirring for a short while the dichloromethane was removed under vacuum and cyclopentadiene (1.2 mmol) added. The resulting mixture was stirred at room temperature for 1 h, after which time the ionic liquid was extracted with diethyl ether (5 × 3 cm3) in air. The organic phase was analysed by 1H NMR spectroscopy and HPLC, and the crude product remaining purified by column chromatography over silica gel (60–200 mesh, 30% ethyl acetate–hexane).Ionic liquid recycle experiments
Following extraction with diethyl ether, the ionic liquid solution was flushed with inert gas and charged with further portions of oxazolidinone 1b (0.16 mmol) and cyclopentadiene (1.2 mmol) at room temperature, and left stirring for a further 1 h.Diels–Alder reactions in the presence of diethyl ether
To a dichloromethane solution (2.0 cm3) of the activated catalyst 7 was added [emim][NTf2] (0.2 cm3) and oxazolidinone 1a or 1b (0.16 mmol). After stirring, the dichloromethane was removed under vacuum and diethyl ether (6.0 cm3) added, followed by cyclopentadiene (1.2 mmol). The resulting biphasic reaction mixture was stirred rapidly under an inert atmosphere for 1 h, after which time the diethyl ether was decanted and analysed by 1H NMR spectroscopy and HPLC.Kinetics
Aliquots of a freshly prepared dichloromethane or [emim][NTf2] solution of the activated catalyst, δ-7a, were taken at regular time intervals at room temperature and quenched with excess (S,S)-1,2-diphenylethylenediamine to afford a diastereotopic mixture. From 31P NMR spectroscopic data, the diastereoisomer ratio was analysed using first order reversible kinetic analysis to yield the rate constant.Results and discussion
Table 1 summarises the results of a comparative study of the Diels–Alder reaction between oxazolidinone 1b and Cp–H in ionic liquids and dichloromethane using catalyst 4a, formed by activation of δ-[(Ph4-NUPHOS)PtCl2] (3a) with Ag[SbF6] (Fig. 3). For each of the ionic liquids studied, significantly higher enantioselectivities were obtained compared with dichloromethane. For example, reaction in [emim][NTf2] gave an ee of 90%, whereas the corresponding reaction in dichloromethane only gave an ee of 67%. While [emim][NTf2] has been chosen for the majority of our studies, catalysts generated in a range of other ionic liquids including [bmim][PF6], [omim][NTf2], and [C4mpyl][NTf2] also performed equally well. It should also be noted that all the ionic liquid reactions were significantly faster than those performed in dichloromethane. Typically, high conversions were achieved in only 1 h at 20 °C, whereas reaction times in excess of 20 h were required to achieve similar levels of conversion in dichloromethane. Although it was possible to achieve high enantioselectivities in dichloromethane with this catalyst, low temperatures and long reaction times were required. Even at −20 °C, the ee of 88% is still slightly lower than that of 93% obtained in ionic liquid at room temperature, which highlights the beneficial influence of ionic liquids over dichloromethane. An enhancement in enantioselectivity was also obtained with catalysts based on the atropisomeric diphosphine BINAP in [emim][NTf2] compared with dichloromethane. For example, under identical conditions, catalyst generated from [{(S)-BINAP}PtCl2] (S-5) and Ag[SbF6] gave an ee of 92% in [emim][NTf2] and only 84% in dichloromethane. As expected catalysts generated from R- and S-[(BINAP)PtCl2] gave the cycloadduct 2b with opposite absolute configurations and comparable enantioselectivities.![Formation of Lewis acid catalysts [(P–P)PtX2].](/image/article/2004/GC/b312761c/b312761c-f3.gif) | ||
| Fig. 3 Formation of Lewis acid catalysts [(P–P)PtX2]. | ||
| Solvent (temp.) | Catalyst | % convad (time) | % endobd | endo eecd (config) |
|---|---|---|---|---|
| a Determined by HPLC/1H NMR spectroscopy.b Endo/exo ratio determined by 1H NMR spectroscopy.c Enantiomeric excess was determined by HPLC (Daicel Chiralcel OD–H).d Average of three runs.e 10 mol%. | ||||
| [emim][NTf2] | δ-4a | 100 (1 h) | 87 | 90 (2R) |
| [bmim][PF6] | δ-4a | 100 (1 h) | 89 | 93 (2R) |
| [omim][NTf2] | δ-4a | 100 (1 h) | 77 | 90 (2R) |
| [C4mpyl][NTf2] | δ-4a | 88 (1 h) | 77 | 90 (2R) |
| [emim][NTf2] | λ-4b | 90 (1 h) | 89 | 91 (2S) |
| CH2Cl2 | δ-4a | 45 (20 h) | 80 | 67 (2R) |
| CH2Cl2 (−20 °C) | δ-4a | 60 (20 h) | 84 | 88 (2R) |
| CH2Cl2 | λ-4b | 22 (20 h) | 88 | 71 (2S) |
| [emim][NTf2] | S-6 | 100 (1 h) | 80 | 92 (2R) |
| [emim][NTf2] | R-6 | 78 (1 h) | 84 | 94 (2S) |
| CH2Cl2 | S-6 | 37 (20 h) | 85 | 84 (2R) |
| [bmim][PF6] | δ-8ae | 69 (1 h) | 89 | 78 (2R) |
| [bmim][NTf2] | δ-8ae | 71 (1 h) | 82 | 90 (2R) |
| [emim][NTf2] | δ-8ae | 93 (1 h) | 88 | 90 (2R) |
| [omim][NTf2] | δ-8ae | 63 (1 h) | 73 | 90 (2R) |
| [C4mpyl][NTf2] | δ-8ae | 86 (1 h) | 76 | 95 (2R) |
| CH2Cl2 | δ-8ae | no reaction | ||
| CH2Cl2 (−20 °C) | δ-8ae | 7 (24 h) | 88 | 93 (2R) |
| [emim][NTf2] | λ-8be | 74 (1 h) | 81 | 91 (2S) |
| [emim][NTf2] | λ-10e | 35 (1 h) | 70 | 85 (2S) |
| CH2Cl2 | λ-10e | no reaction | ||
We are confident that the enhancement in the ee observed with catalysts based on Ph4-NUPHOS in the ionic liquids does not simply reflect the increased rate of Diels–Alder reaction in the two solvents since the rate of racemisation is also decreased significantly in ionic liquids. 31P NMR data acquired after quenching the catalyst with S,S-dpeda gave a first order racemisation rate constant of 9 × 10−6 s−1 in dichloromethane whereas no racemisation occurred in [emim][NTf2] within the NMR detection limit. However, there also appears to be an enhancement in enantioselectivity with the atropisomeric catalyst, δ-6, albeit smaller than that found for catalysts based on conformationally flexible ligands, indicating that the ionic liquid has an intrinsic effect on the ee.
As noted above, the Lewis acid fragments δ/λ-(R4-NUPHOS)Pt(OTf)2 (8a/b), generated by protonation of δ/λ-(R4-NUPHOS)Pt{(S)-BINOL} (7a/b) with triflic acid, (Fig. 3) are highly effective catalysts for the Diels–Alder reaction between oxazolidinone 1b and Cp–H at −20 °C. However, it has not been possible to perform the Diels–Alder reaction with oxazolidinone 1b at 20 °C in dichloromethane due to the instability of the catalyst with respect to cyclopentadiene. Even addition of diene to a dichloromethane solution of catalyst δ-8a at −78 °C and subsequent warming to 20 °C resulted in the rapid appearance of a characteristic deep blue–green coloration and after 1 h conversions were less than 5%. 31P NMR analysis of this reaction mixture revealed a singlet at δ −5.7 flanked by platinum satellites (1JPt–P = 4542 Hz) which corresponds to the catalytically inactive cation [(Ph4-NUPHOS)Pt(η5-Cp)]+, based on the similarity of its spectroscopic data with that of [(BIPHEP)Pt(η5-Cp)]+, reported earlier by Becker et al.4 This species does not form in ionic liquids and the reaction proceeds with high enantioselectivity at room temperature giving an ee of 90% and high conversions. Moreover, a comparison of the data in Table 1 clearly shows that the performance of catalysts generated from δ-(Ph4-NUPHOS)Pt{(S)-BINOL}(δ-7a) are comparable to those generated by halide abstraction from the corresponding dichloride complex δ-(Ph4-NUPHOS)PtCl2 (δ-3a). It should be noted that the Diels–Alder reaction does proceed in dichloromethane at or below −20 °C using the catalyst generated from δ-(Ph4-NUPHOS)Pt{(S)-BINOL}(δ-7a). However, the ee of 93% obtained at −20 °C is only comparable with that obtained in ionic liquid at room temperature. At this stage we believe that the difference between the dichloromethane and the ionic liquids is due to the stability of catalyst generated from the BINOLate precursors δ/λ-7a/b and not to a difference in stability of the [(Ph4-NUPHOS)Pt(η5-Cp)]+ cation, which has been independently prepared and shown to be stable but inactive in [emim][NTf2]; a catalytic reaction giving less than 2% conversion at 20 °C. Similarly, catalyst mixtures generated from λ-(Me4-NUPHOS)Pt{(S)-BINOL} (λ-7b) in [emim][NTf2] show high enantioselectivity for the Diels–Alder reaction at room temperature, giving an ee of 91%, but no reaction in dichloromethane. Not surprisingly, catalyst generated from λ-(BIPHEP)Pt{(S)-BINOL} (λ-9) also showed comparable behaviour in ionic liquids at 20 °C but again little conversion in dichloromethane due to rapid formation of [(BIPHEP)Pt(η5-Cp)]+.
Whilst it is not straightforward to recycle the catalyst in dichloromethane, simple extraction with diethyl ether allowed the ionic liquid systems to recycle efficiently. Table 2 summarises a series of recycle experiments using δ-[(Ph4-NUPHOS)Pt(SbF6)2] (δ-4a) and [{(S)-BINAP}Pt(SbF6)2] (S-6) and δ-[(Ph4-NUPHOS)Pt(OTf)2] (δ-8a) in [emim][NTf2], and δ-[(Ph4-NUPHOS)Pt(SbF6)2] (δ-4a) in [bmim][PF6]. The extractions were performed in air and no significant change in the ee was found for successive reactions. In general, the catalyst–ionic liquid systems tested showed only a small decrease in activity on each recycle. ICP analysis of the ether extract showed that the platinum leaching was below the detection limit and therefore it is possible that this reduction is due to the gradual build up of inactive platinum complexes, possibly based on the cyclopentadiene.
| Ionic liquid | Catalyst (mol%) | Run: % convad | % endobd | endo eecd (config) |
|---|---|---|---|---|
| a Determined by HPLC/1H NMR. All reactions run for 1hb Determined by 1H NMR spectroscopy.c Enantiomeric excess was determined by HPLC (Daicel Chiralcel OD–H).d Obtained from single run only. | ||||
| [bmim][PF6] | δ-4a (20) | 1: 100 | 89 | 93 (2R) |
| 2: 96 | 87 | 90 (2R) | ||
| 3: 90 | 84 | 90 (2R) | ||
| [emim][NTf2] | δ-4a (20) | 1: 100 | 87 | 90 (2R) |
| 2: 100 | 85 | 91 (2R) | ||
| 3: 96 | 85 | 89 (2R) | ||
| [emim][NTf2] | S-6 (20) | 1: 100 | 80 | 92 (2R) |
| 2: 100 | 69 | 94 (2R) | ||
| 3: 96 | 82 | 91 (2R) | ||
| [emim][NTf2] | δ-8a (10) | 1: 71 | 82 | 90 (2R) |
| 2: 64 | 79 | 88 (2R) | ||
| 3: 65 | 79 | 89 (2R) | ||
Preliminary data have also shown that there is potential for the ionic liquid process to be scaled up using a continuous extraction and only a nominal volume of ionic liquid. Reactions performed using 10 mol% of the catalyst system generated from δ/λ-[(R4-NUPHOS)Pt{(S)-BINOL}] (R = Ph, 7a; R = Me, 7b) in 0.2 cm3 of [emim][NTf2] (compared with 2.0 cm3 for the reactions described above) with 6.0 cm3 diethyl ether as an immiscible co-solvent for extraction showed slower reaction kinetics but with an increased ee, 96% vs. 90% (Fig. 4). As found for reactions in the absence of diethyl ether, good recyclability is found in the biphasic solvent system.
![Variation in %conversion (closed symbols) and %ee
(open symbols) for the Diels–Alder reaction between 1b and Cp–H catalyzed by δ-8a
(10 mol%) in [emim][NTf2]
(circles) and [emim][NTf2]–diethyl ether (squares) with respect to time at 20 °C.](/image/article/2004/GC/b312761c/b312761c-f4.gif) | ||
| Fig. 4 Variation in %conversion (closed symbols) and %ee (open symbols) for the Diels–Alder reaction between 1b and Cp–H catalyzed by δ-8a (10 mol%) in [emim][NTf2] (circles) and [emim][NTf2]–diethyl ether (squares) with respect to time at 20 °C. | ||
In order to obtain high ee's for the Diels–Alder reaction between oxazolidinone 1a and Cp–H using the catalyst system generated from δ-[(Ph4-NUPHOS)Pt{(S)-BINOL}] (δ-7a), the reactions had to be performed at −20 °C or below. Unfortunately, ionic liquids have relatively high viscosity below 0 °C and this in turn can result in significantly reduced reaction rates. This was overcome by performing these reactions in ionic liquid–diethyl ether mixtures, which increased the stirring efficiency compared with the pure ionic liquid and resulted in good conversions. As with the reactions described above between 1b and Cp–H, the ionic liquid increased both the reaction rate and ee compared with CH2Cl2. Fig. 5 shows the variation in conversion and ee at −20 °C and −40 °C in [emim][NTf2]–diethyl ether and CH2Cl2, respectively. Clearly the rate is much higher in the mixed solvent system compared with CH2Cl2. Although this could be due to the difference in reaction temperature, it should be noted that at −20 °C, only 45% conversion (48% ee) was obtained in CH2Cl2 over 24 hours due to the slow formation of the inactive [(Ph4-NUPHOS)Pt(η5-Cp)]+ cation compared with 99% conversion (93% ee) after 2 h in [emim][NTf2]–diethyl ether. In contrast, the ee normally increases with decreasing temperature; however, it is evident that, despite the higher temperature, the ionic liquid–diethyl ether system results in a significantly higher ee than found in CH2Cl2 at all conversions. Unlike in the ionic liquid–diethyl ether system, the ee is also found to decrease as the reaction proceeds in CH2Cl2. As found for reactions in the absence of diethyl ether, good recyclability is found in the biphasic solvent system.
![Variation in %conversion (closed symbols) and %ee
(open symbols) for the Diels–Alder reaction between 1a and Cp–H catalyzed by δ-8a
(20 mol%) in [emim][NTf2]–diethyl ether at −20 °C (squares) and in dichloromethane at −40 °C (circles) with respect to time.](/image/article/2004/GC/b312761c/b312761c-f5.gif) | ||
| Fig. 5 Variation in %conversion (closed symbols) and %ee (open symbols) for the Diels–Alder reaction between 1a and Cp–H catalyzed by δ-8a (20 mol%) in [emim][NTf2]–diethyl ether at −20 °C (squares) and in dichloromethane at −40 °C (circles) with respect to time. | ||
In conclusion, we have demonstrated that ionic liquid can not only significantly increase the rate of Diels–Alder reactions and the recyclability of the catalyst but also allow high enantioselectivities to be achieved without the need to recourse to low temperatures. This coupled with the use of conformationally flexible inexpensive ligands and the ability to perform semi-continuous reactions allows the possibility of scale-up. Ionic liquids also have the potential to replace chlorinated hydrocarbons often used for this type of Diels–Alder reaction.
Acknowledgements
We acknowledge support from the EPSRC under grants GR/R42078 and GR/R42061 and H. L. acknowledges support from the McClay Trust, QUB.References
- (a) K. Mikami, M. Terada, T. Korenaga, Y. Matsumoto and S. Matsukawa, Acc. Chem. Res., 2000, 33, 391 CrossRef CAS; (b) K. Mikami, M. Terada, T. Korenaga, Y. Matsumoto, M. Ueki and R. Angeland, Angew. Chem., Int. Ed., 2000, 39, 3532 CrossRef CAS.
- K. Mikami and S. Matsukawa, Nature, 1997, 385, 613 CrossRef CAS.
- S. Matsukawa and K. Mikami, Tetrahedron: Asymmetry, 1997, 8, 815 CrossRef CAS.
- J. J. Becker, P. S. White and M. R. Gagné, J. Am. Chem. Soc., 2001, 123, 9478 CrossRef CAS.
- S. Doherty, J. G. Knight, E. G. Robins, T. H. Scanlan, P. A. Champkin and W. Clegg, J. Am. Chem. Soc., 2001, 123, 5110 CrossRef CAS.
- (a) S. Doherty, E. G. Robins, M. Nieuwenhuyzen, J. G. Knight, P. A. Champkin and W. Clegg, Organometallics, 2002, 21, 1383 CrossRef CAS; (b) S. Doherty, C. R. Newman, C. Hardacre, M. Nieuwenhuyzen and J. G. Knight, Organometallics, 2003, 22, 1452 CrossRef CAS.
- S. Doherty, C. R. Newman, R. K. Rath, H.-K. Luo, M. Nieuwenhuyzen and J. G. Knight, Org. Lett., 2003, 5, 3863 CrossRef CAS.
- M. J. Earle, P. B. McCormac and K. R. Seddon, Green Chem., 1999, 1, 23 RSC.
- C. E. Song, W. H. Shim, E. J. Roh and J. H. Chio, Chem. Commun., 2000, 1695 RSC.
- C. J. Mathews, P. J. Smith and T. Welton, Chem. Commun., 2000, 1249 RSC.
- A. J. Carmichael, M. J. Earle, J. D. Holbrey, P. B. McCormac and K. R. Seddon, Org. Lett., 1999, 1, 997 CrossRef CAS.
- C. deBellefon, E. Pollet and P. Grenouillet, J. Mol. Catal., 1999, 145, 121 Search PubMed.
- A. J. Carmichael, D. M. Haddleton, S. A. F. Bon and K. R. Seddon, Chem. Commun., 2000, 1237 RSC.
- M. F. Pinheiro, R. S. Mauler and R. F. deSouza, Macromol. Rapid Commun., 2001, 22, 425 CrossRef CAS.
- C. Hardacre, J. D. Holbrey, S. P. Katdare and K. R. Seddon, Green Chem., 2002, 4, 143 RSC.
- Y. Chauvin, L. Mussman and H. Olivier, Angew. Chem., Int. Ed. Engl., 1996, 34, 2698 CrossRef.
- P. J. Dyson, D. J. Ellis and T. Welton, Can. J. Chem., 2001, 79, 705 CrossRef CAS; P. J. Dyson, D. J. Ellis, D. G. Parker and T. Welton, Chem. Commun., 1999, 25 RSC.
- K. Anderson, P. Goodrich, C. Hardacre and D. W. Rooney, Green Chem., 2003, 5, 448 RSC.
- D. Zim, R. F. de Souza, J. Dupont and A. L. Monteiro, Tetrahedron Lett., 1998, 39, 7071 CrossRef CAS.
- Ionic Liquids in Synthesis, ed. P. Wasserscheid and T. Welton, Wiley-VCH, Weinheim, 2003 Search PubMed.
- C. E. Song and E. J. Roh, Chem. Commun., 2000, 837 RSC.
- C. E. Song, C. R. Oh, E. J. Roh and D. J. Choo, Chem. Commun., 2000, 1743 RSC.
- S. Guernik, A. Wolfson, M. Herskowitz, N. Greenspoon and S. Geresh, Chem. Commun., 2001, 2314 RSC.
- (a) L. C. Branco and C. A. M. Afonso, Chem. Commun., 2002, 3036 RSC; (b) C. E. Song, D. Jung, E. J. Roh, S. Lee and D. Y. Chi, Chem. Commun., 2002, 3038 RSC.
- C. R. Oh, D. J. Choo, W. H. Shim, D. H. Lee, E. J. Roh, S. Leec and C. E. Song, Chem. Commun., 2003, 1100 RSC.
- P. G. Jessop, R. R. Stanley, R. A. Brown, C. A. Eckert, C. L. Liotta, T. T. Ngo and P. Pollet, Green Chem., 2003, 5, 123 RSC.
- C. Baudequin, J. Baudoux, J. Levillain, D. Cahard, A.-C. Gaumont and J.-C. Plaquevent, Tetrahedron Asymmetry, 2003, 14, 3081 CrossRef CAS.
- I. Meracz and T. Oh, Tetrahedron Lett., 2003, 44, 6465 CrossRef CAS.
- P. Bonhôte, A. P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168 CrossRef CAS.
- (a) J. D. Holbrey and K. R. Seddon, J. Chem. Soc., Dalton Trans., 1999, 2133 RSC; (b) C. M. Gordon, J. D. Holbrey, A. R. Kennedy and K. R. Seddon, J. Mater. Chem., 1998, 8, 2627 RSC.
- D. R. MacFarlane, P. Meakin, J. Sun, N. Amini and M. Forsyth, J. Phys. Chem. B, 1999, 103, 4164 CrossRef CAS.
- P. Gugger, S. O. Limmer, A. A. Watson, A. C. Willis and S. B. Wild, Inorg. Chem., 1993, 32, 5692 CrossRef CAS.
- M. D. Tudor, J. J. Becker, P. S. White and M. R. Gagné, Organometallics, 2000, 19, 4376 CrossRef CAS.
- D. A. Evans, K. T. Chapman and J. Bisaha, J. Am. Chem. Soc., 1988, 110, 1238 CrossRef CAS.
- A. K. Ghosh and H. Matsuda, Org. Lett., 1999, 1, 2157 CrossRef.
| This journal is © The Royal Society of Chemistry 2004 |