Kinetic studies for processes of liquid-phase alkylation of aromatics over solid acid catalysts
Paolo Beltrame* and Giovanni Zuretti
Dipartimento di Chimica Fisica ed Elettrochimica, Università degli Studi di Milano, Via Golgi 19, I-20133 Milano, Italy. E-mail: paolo.beltrame@unimi.it; Fax: +39-02-50314300
First published on 22nd October 2003
Abstract
The liquid-phase alkylation of aromatics has been effected traditionally with catalysis by inorganic acids or AlCl3 and similar compounds. However the increasing importance of solid acid catalysts is changing the procedures in this field. One of the prerequisites for the development of relevant processes, and the design of reactors, is knowledge of the kinetic data in reactions catalyzed by solid acids. This review examines the information available on the kinetics of alkylations catalyzed by solid acids, mainly zeolites, supported metal chlorides and oxides, mesoporous (MCM) materials, and heteropolyacids, but also other less studied materials.
 Paolo Beltrame Paolo Beltrame | Paolo Beltrame was born in Milano, Italy, in 1930. He received his “Laurea” in Industrial Chemistry from the University of Milano, in 1954. In 1955 he joined the same University as assistant professor. He carried out research work at University College London, UK, in 1959, and at University of California at Los Angeles, USA, in 1962. In 1970 he joined the University of Cagliari, Italy, as full professor. Currently he is full professor at the University of Milano, for Industrial Chemistry curricula. His current research interests are chemical kinetics and catalysis of reactions of industrial importance. |
| The alkylation of aromatics is a particularly important area of industrial chemistry. In searching for more environmentally benign replacements for traditional hazardous acid catalysts such as aluminium chloride, numerous solid acid catalysts have been suggested. Determining the most appropriate of these is very difficult and here the information available on the kinetics of these reactions is renewed in an attempt to help progress the development of new greener processes. JHC |
Introduction
The alkylation of aromatic compounds with olefins, halides or alcohols, traditionally catalyzed by the highly corrosive inorganic Brønsted and Lewis acids, can be carried out over solid acid catalysts, the advantages being easier handling, less equipment corrosion, few problems with regard to environmental disposal and toxicity, simple product separation, and catalyst recycling.The mechanism proposed for these reactions is the general one for electrophilic aromatic substitution. In particular, it is proposed that the alkylating agent gives rise to an adsorbed organic cation. This may happen by protonation over protonating acids, producing, for instance, (C6H5CH2+)ads from benzyl chloride1 or (C6H11+)ads from cyclohexene2 over a heteropolyacid, or by homolytic redox mechanism producing the cation from benzyl chloride over supported metallic oxides or chlorides such as Fe3+ chloride.3,4 The next step is the reaction of the adsorbed cation with the aromatic reagent, present either in the fluid phase or itself adsorbed on the catalytic surface.
Our aim is to review liquid-phase alkylations over solid acid catalysts from the viewpoint of reaction kinetics. The articles on the subject found in the literature have been classified into four categories:
a) work including a discussion on mass transfer resistances and their weight in the process (the conclusions drawn are that such resistances are negligible), followed by a search for kinetic equations and an evaluation of the relevant parameters. The results in these cases can be reasonably considered as pertaining to intrinsic kinetics;
b) kinetic studies similar to those in (a), apart from the discussion on mass transfer resistances;
c) studies including the measurement of reaction rates, without a search for kinetic equations;
d) other articles containing kinetic data, although of less straightforward interpretation.
From the point of view of chemical engineering, that requires intrinsic kinetic equations, the first two categories are the most useful, although the results of type (b) may not be as reliable as those of type (a).
The reactions are usually carried out with an excess of aromatic reagent with respect to the alkylating agent, and in many cases it is the aromatic itself that is the reaction solvent; however in some cases there can be a different (usually non-aromatic) solvent.
This literature review has revealed great variety in the approaches to kinetic measurement, so that, given the differences in meaning of the parameters, it is not easy to make comparisons. Thus it has been considered important to unify, as far as possible, parameter definitions along the following lines.
Some authors give pseudo-first order rate coefficients with a dimension time−1, while others give them with dimensions volume·mass−1·time−1. We have chosen to take eqn. (1) as a typical first-order rate equation:
| r = k1·Calk·Ccat | (1) |
Other authors have chosen to give second-order rate coefficients. In these cases, we have chosen a kinetic equation for monoalkylation such as eqn. (2):
| r = k2·Carom·Calk·Ccat | (2) |
In some cases, kinetic equations of the Langmuir–Hinshelwood type have been proposed. For monoalkylation such equations have the form of eqn. (3), i.e., they contain a numerator similar to eqn. (2) but also a denominator which includes one or more adsorption constants Kads for one or more components of the reaction system.
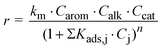 | (3) |
Analogous equations are employed for dialkylations and other reactions. It should be noted that in these equations the coefficient km is not a “true” rate coefficient because it also includes adsorption coefficients.
Finally, some authors were concerned about the deactivation rate of their catalysts, leading them to also study the kinetics of this phenomenon and to provide deactivation rate coefficients, with dimension time−1. We have given such coefficients as kd (min−1). It is worth noting that they appear as first-order coefficients, but the relevant equations usually employ dimensionless variables so that the dimension of kd does not give an idea of the actual form that the kinetic equation takes. Such forms will be specified when mentioning these deactivation coefficients.
In the cases of type (c), the measured rates given by the authors have been converted to units mmol g−1·min−1, for easier comparison.
As to the equipment for kinetic measurement, most articles mention stirred batch reactors, obviously slurry reactors, given the presence of solid catalyst. In some cases, when the nature of the alkylating agent and the reaction temperature required pressures higher than ambient pressure, stirred autoclaves were used. With gaseous alkylating agents, a semi-batch reactor was sometimes employed, maintaining a known Calk value in the system.
2 Kinetic equations and kinetic parameters
Studies of type (a) were carried out at high stirring speed, checking, in most cases, that a further enhancement of the speed had no effect on the kinetics. In this way significant external mass transfer resistance was excluded. With regard to intraparticle resistance, care was taken (in most cases) to use very fine catalyst particles; in such cases very low Thiele modulus values and therefore catalyst effectiveness values close to unity can be calculated.Table 1 shows typical results of studies of this type. The catalysts presented by the papers mentioned in Table 1 are zeolites (microporous crystalline aluminosilicates),17 amorphous silica-aluminas, supported metal oxides, HAlMCM-41 (mesoporous aluminosilicates presenting uniform channels from 15 to 100 Å in size),18,19 sulfated zirconia supported on HMS (a mesoporous material similar to MCM-41),20 and Nafion (a perfluorinated polymeric sulfonic acid)21 in the form of a Nafion–Silica composite.22
| Reference | Aromatic (solvent)a | Alkylating agent (pressure)b | Reactor [rpm]c | T/°C | Catalyst | Ccat/g l−1 | Parameter values (at temp. T,°C) |
|---|---|---|---|---|---|---|---|
| a If different from the aromatic itself.b If different from ambient pressure (1 atm = 101.325 KPa).c Stirrer speed, when specified.d Benzyl chloride.e For an equation −da/dt = kda2, where a is the catalyst activity coefficient.f Methyl-tert-butyl ether, as a source of isobutene.g Reaction carried out in a continuous tubular reactor.h km1 and km2 correspond to the monobenzylation to 1-benzyl- and 2-benzyl-naphthalene, respectively.i Benzyl alcohol. | |||||||
| Pseudo-first-order kinetic equation (k1/l g−1 min−1) (kd/min−1) | |||||||
| 5 | phenol | c-hexene | batch | 100 | HY, Si/Al = 25 | ||
| (dichlorobenzene) | [1000] | 1.4 | k1 = 0.0125 | ||||
| (cyclohexane) | 1.5 | k1 = 0.0056 | |||||
| (nitropropane) | 1.7 | k1 = 0.0011 | |||||
| 6 | biphenyl | BzCld | batch | 80 | HY, Si/Al = 7 | 5–13 | k1 = 0.0010 |
| (cyclohexane) | [825] | kd = 0.25e | |||||
| 7 | benzene | BzCl | batch | 60–80 | In2O3/Hβ, Si/Al = 27 | 7.1 | k1 = 0.090 (80) |
| anisole | [high speed + N2 flux] | 80 | k1 = 0.014 | ||||
| Second-order kinetic equation (k2/l2 mol−1 g−1 min−1) | |||||||
| 8 | p-cresol | MTBEf | autoclave | 80–115 | sulfated ZrO2/HMS | 30.8 | k2 = 0.000020 (100) |
| (>1 atm) | [700] | ||||||
| 9,10 | benzene | 1-dodecene | fixed bed | 85–125 | H3PW12O40/SiO2 | g | k2 = 0.000012 (100) |
| (24 atm) | |||||||
| Second-order kinetic equation (k2/mol g−1 min−1 (mol fraction)−2) | |||||||
| 11 | benzene | propene | fixed bed | 200–240 | CaHY | g | k2 = 0.0024 (230) |
| (40 atm) | |||||||
| Langmuir–Hinshelwood kinetic model (km/l2 mol−1 g−1 min−1) (Kads/l mol−1) | |||||||
| 12,13 | biphenyl | BzCl | batch | 80 | amorphous | 8–16 | km = 0.00058 |
| (cyclohexane) | [825] | silica-alumina, Si/Al = 6.2 | KBzCl = 10.6 (n = 1) | ||||
| 13 | biphenyl | BzCl | batch | 80 | amorphous | 5–16 | km = 0.0021 |
| (cyclohexane) | [825] | silica-alumina, Si/Al = 2.7 | KBzCl = 12.2 (n = 1) | ||||
| biphenyl | BzCl | batch | 80 | HAlMCM-41, Si/Al = 19 | 3 | km = 0.0118 | |
| (cyclohexane) | [825] | KBzCl = 6.53 (n = 1) | |||||
| 14 | biphenyl | BzCl | batch | 80 | HAlMCM-41, Si/Al = 19 | 2–4 | km = 0.0714 |
| (cyclohexane) | [825] | KBzCl = 24.3 | |||||
| KBIP = 5.4 (n = 2) | |||||||
| kd < 0.001e | |||||||
| 15 | naphthalene | BzCl | batch | 80 | HAlMCM-41, Si/Al = 19.5 | 5 | km1 = 0.46h |
| (cyclohexane) | [1000 + N2 flux] | km2 = 0.19h | |||||
| KBzCl = 10.3 | |||||||
| KNA = 12.6 (n = 2) | |||||||
| 16 | naphthalene | BzOHi | batch | 80 | Nafion-Silica | 2–3 | km = 0.50 |
| (cyclohexane) | [1000 + N2 flux] | composite (15%) | KBzOH = 110 (n = 2) | ||||
The alkylating agents in Table 1 are, in many cases, benzyl chloride or benzyl alcohol, and, less commonly, olefins. Most of the studies were carried out at relatively low temperatures, in the 60–100 °C range, with catalyst doses of a few grams per liter.
Table 2 shows typical results of studies of type (b). The catalysts concerned include, besides the kind of solid acids already present in Table 1, K10 (an acid-treated montmorillonite clay of large surface area), Clayzic (zinc chloride supported on K10 montmorillonite),33 a heteropolyacid H0.5Cs2.5PW12O40, and supported metal chlorides.
| Reference | Aromatic (solvent)a | Alkylating agent (pressure)b | Reactor [rpm]c | T/°C | Catalyst | Ccat/g l−1 | Parameter values (at temp. T, °C) |
|---|---|---|---|---|---|---|---|
| a See the corresponding footnote in Table 1.b See the corresponding footnote in Table 1.c See the corresponding footnote in Table 1.d See the corresponding footnote in Table 1.e Thermally activated.f Unspecified.g For an equation −da/dt = kda3, where a is the catalyst activity coefficient.h Benzyl alcohol.i With N2 flux. | |||||||
| Pseudo-first-order kinetic equation (k1/l g−1 min−1) (kd/min−1) | |||||||
| 23 | anisole | BzCld | batch | 20–50 | K10 | 30.3 | k1 = 0.000043 (50) |
| benzene | BzCl | batch | 20–60 | clayzic | 35.7 | k1 = 0.000104 (50) | |
| (unactivated) | |||||||
| 24 | benzene | BzCl | batch | 40 | clayzice | 35.7 | k1 = 0.00095 |
| chlorobenzene | BzCl | batch | 40 | clayzice | 32.3 | k1 = 0.00068 | |
| 25 | toluene | BzCl | batch | 40 | clayzice | 30.8 | k1 = 0.0319 |
| ethylbenzene | BzCl | batch | 40 | clayzice | 30.8 | k1 = 0.0015 | |
| 26,27 | benzene | 1-dodecene | fluidized bed | 70–90 | HY, Si/Al = 1.6 | f | k1 = 1.12 (80) |
| kd = 0.31g | |||||||
| 1 | benzene | BzCl | batch | 80 | H0.5Cs2.5PW12O40 | 174 | k1 = 0.027 |
| 28 | benzene | 1-dodecene | batch | 100–140 | HY, Si/Al = 2.5 | 5 | k1 = 0.0049 (100) |
| (decane) | (6–9 atm) | [500] | |||||
| 29 | toluene | BzOHh | batch | 90 | HAlMCM-41, Si/Al = ca. 10 | 8.7 | k1 = 0.00017 |
| [+air flux] | |||||||
| 30 | benzene | BzCl | batchi | 80 | TlOx20%/ZrO2 | 7.1 | k1 = 0.0411 |
| (low surface) | |||||||
| anisole | BzCl | batchi | 80 | TlOx20%/ZrO2 | 7.1 | k1 = 0.0287 | |
| (low surface) | |||||||
| 31 | benzene | BzCl | batchi | 60–80 | Ga2O3/HZSM-5, Si/Al = 31 | 7.1 | k1 = 0.0210 (80) |
| 32 | benzene | BzCl | batchi | 60–80 | InCl3/K10 | 7.1 | k1 = 0.0737 (80) |
| benzene | BzCl | batchi | 60–80 | GaCl3/K10 | 7.1 | k1 = 0.0451 (80) | |
| benzene | BzCl | batchi | 60–80 | FeCl3/K10 | 7.1 | k1 = 0.0772 (80) | |
| toluene | BzCl | batchi | 80 | InCl3/K10 | 7.1 | k1 = 0.0592 | |
| mesitylene | BzCl | batchi | 80 | InCl3/K10 | 7.1 | k1 = 0.0521 | |
The most common alkylating agent present in Table 2 is again benzyl chloride, benzyl alcohol and olefins being less commonly employed. The reaction temperatures were low, even lower than those of Table 1.
The values shown in Tables 1 and 2 concern several different reactions, a fact that makes comparison difficult. The kinetic measurements (pseudo-first order coefficients) on benzene alkylation using benzyl chloride, carried out at 80 °C or at temperatures close to 80 °C, provide the largest data set, allowing comparison of the different catalysts. The order of decreasing catalytic activity appears to be the following:
| In2O3/Hβ7 > FeCl3/K10, InCl3/K1032 > GaCl3/K1032 > TlOx/ZrO230 > H0.5Cs2.5PW12O401 > Ga2O3/HZSM–531 > Clayzic24 |
A few reactions have been carried out using a solvent different from the aromatic reagent. Eight different solvents were used in the reaction of phenol with cyclohexene over HY zeolite (Table 1), revealing that the alkylation rate had a peak for solvents of medium polarity.5 Since the measured parameter was a pseudo-first order coefficient with respect to Calk, it could be expected that these reactions, having a lower Carom value, would show lower k1-values than reactions carried out with an aromatic excess, the excess acting as both reagent and solvent. Further examples are given by the reactions of biphenyl with benzyl chloride in cyclohexane6 (Table 1), and benzene with 1-dodecene in decane28 (Table 2); both reactions employ a HY zeolite catalyst.
Among the cases listed in Table 2, some values are noteworthy: it is surprising how slow the reaction of toluene with benzyl alcohol over MCM-4129 is, and how fast is that of benzene with 1–dodecene over HY;26,27 in the latter case, the reason could be that the authors used a particularly efficient reactor.
Table 2 offers, for the same catalyst (Clayzic), the opportunity to compare an unactivated sample with a thermally activated one, under the same conditions:23,24 activation increased the rate by more than one order of magnitude. The kinetic effect of alkyl substituents on benzene is less clear: with Clayzic as the catalyst, toluene and ethylbenzene reacted faster than benzene,24,25 while with InCl3/K10 as catalyst, toluene and mesitylene were found to be slightly less reactive than benzene.32
One group of studies (Table 1) employed the kinetic model corresponding to eqn. (3). In these, no straightforward comparison of the different reactions can be made as the rates depend not only on the coefficients km in the numerator but also on the constants Kads,j and the value of n in the denominator of the kinetic equations. However, it is possible to see that for the benzylation of biphenyl, the high-alumina silico-aluminate catalyst (Si/Al = 2.7) is more active than the low-alumina catalyst (Si/Al = 6.2),13 and catalyst HAlMCM-41 is more active than either of the previous ones.13,14 For the naphthalene reactions, data for the same temperature and the same solvent are available, but different benzylating agents and different catalysts were used, so the figures in Table 2 cannot be compared directly. However, it has been reported that BzOH is much more reactive than BzCl on the Nafion-silica composite, while the chloride is more reactive than the alcohol on the MCM-41 catalyst.16
3 Other kinetic measurements
Table 3 gives results typical of the type (c) studies, where the catalysts of interest include, besides the kind of solid acids seen in the previous paragraph, also an ion-exchange resin (Amberlyst 15) and SAPO–11 (a component of a class of microporous silicoaluminophosphates).45 The alkylating agents are benzyl chloride and benzyl alcohol, olefins, and adamantyl bromide. Note that the temperatures are often higher than those in Tables 1 and 2.| Reference | Aromatic (solvent)a | Alkylating agent (pressure)b | Reactor [rpm]c | T/°C | Catalyst | r/mmol g−1 min−1 |
|---|---|---|---|---|---|---|
| a See the corresponding footnote in Table 1.b See the corresponding footnote in Table 1.c See the corresponding footnote in Table 1.d See the corresponding footnote in Table 1.e With catalyst deactivation due to olefin oligomers (coke); for a zero-order reaction of formation of % coke, measured kd = 0.056 min−1 (80 °C).f Benzyl alcohol.g Adamantyl bromide. | ||||||
| 34 | benzene | BzCld | batch | 95 | sulfated ZrO2 | r = 0.194 |
| [900] | ||||||
| 2 | m-xylene | c-hexene | batch | 100 | H0.5Cs2.5PW12O40 | r = 0.41 |
| mesitylene | c-hexene | batch | 100 | H0.5Cs2.5PW12O40 | r = 0.44 | |
| 35 | toluene | BzCl | batch | 110 | HY, Si/Al = 20 | r = 1.12 |
| [700 + N2 flux] | ||||||
| 36 | benzene | 1-dodecene | batch | 100–130 | HY, Si/Al = 2.7 | r = 2.80 (100) |
| (decane) | (6 atm) | [500] | ||||
| 37 | o-xylene | styrene | batch | 60–100 | Amberlyst 15e | r = 0.49 (80) |
| [1500] | ||||||
| 38 | biphenyl | propene | stirred | 250–300 | SAPO-11 | r = 0.015 (300) |
| (decalin) | (>1 atm) | autoclave | ||||
| biphenyl | propene | stirred | 250 | H-Mordenite | r = 0.055 | |
| (decalin) | (>1 atm) | autoclave | ||||
| 39 | benzene | BzOHf | batch | 80 | Nafion–Silica | r = 1.72 |
| [+N2 flux] | composite (13%) | |||||
| p-xylene | BzOH | batch | 100 | Nafion–Silica | r = 2.97 | |
| [+N2 flux] | composite (13%) | |||||
| 40 | naphthalene | BzCl | batch | 40–70 | Hβ, Si/Al = 13 | r = 0.009 (70) |
| (dichloroethane) | ||||||
| 41 | o-xylene | BzCl | batch | 90–135 | Hβ, Si/Al = 13 | r = 0.21 (90) |
| 42 | toluene | Ad-Brg | batch | 111 | Nafion–Silica | r = 3.38 |
| composite (13%) | ||||||
| 43 | benzene | 1-dodecene | batch | 80 | Nafion–Silica | r = 0.080 |
| comp. (13%) [Type 1] | ||||||
| benzene | 1-dodecene | batch | 80 | Nafion–Silica | r = 1.08 | |
| comp. (13%) [Type 2] | ||||||
| benzene | propene | semi-batch | 70 | Nafion–Silica | r = 0.115 | |
| comp. (13%) [Type 1] | ||||||
| benzene | propene | semi-batch | 70 | Nafion–Silica | r = 0.72 | |
| comp. (13%) [Type 2] | ||||||
| 44 | bromobenzene | BzCl | batch | 130 | HY, Si/Al = 2.8 | r = 0.50 |
| bromobenzene | BzCl | batch | 130 | Hβ, Si/Al = 4.5 | r = 0.40 | |
| bromobenzene | BzCl | batch | 130 | Hβ, Si/Al = 13 | r = 0.23 | |
| bromobenzene | BzCl | batch | 130 | K10, Si/Al = 3.1 | r = 0.62 | |
| 4 | benzene | BzCl | batch | 80 | FeCl3/K10 | r = 24.2 |
| [+N2 flux] | ||||||
| benzene | BzCl | batch | 80 | FeCl3/Si-MCM41 | r = 19.8 | |
| [+N2 flux] | ||||||
The reactions considered in Table 3 were carried out at different temperatures and almost all differ one from the other, making it hard to compare the reaction rates. However for a common temperature an evaluation reveals the reaction rates of benzyl chloride with benzene and substituted benzenes to be similar (within an order of magnitude), apart from the case of the benzene reactions over supported FeCl3 catalysts, where the rates were exceptionally high.4 Again, the lowest rates were measured for reactions in solutions having low values of Carom, as for biphenyl + propene in decalin38 and for naphthalene + BzCl in dichloroethane.40
Needless to say, catalyst preparation matters. Table 3 shows two reactions of benzene with olefins,43 where two types of Nafion–silica composite can be compared and it can be seen that they are of quite different activity, Type 2 being an order of magnitude more active than Type 1 for the same reaction at the same temperature.
In addition to the papers in Tables 1–3, other articles [type (d)] deal with the use of a heteropolyacid supported on silica,46 Amberlyst 15 and analogous resins,47–51 Clayzic,52 sulfated zirconia,53–54 MCM–22,55 zeolite Hβ,56 and heteropolyacid supported on K10 clay.57–59
The rates mentioned are those of forward alkylation reactions, as most authors gave no consideration to backward reactions, and those who did take them into account, for the alkylation of p-cresol with isobutene51 and that of benzene with propene,56 found them to be negligible.
The values of the kinetic parameters (coefficients, adsorption constants, rates) given in Tables 1–3 are ascribed to a specified temperature. However, some kinetic measurements were carried out at different temperatures, and apparent activation energy values (EA) have been reported.
Table 4 shows the EA values available for the reactions mentioned in the previous Tables. They are presented separately for the reactions of Table 1 and 2 (in both cases mostly values from 10 to 20 kcal mol−1), and Table 3. In the case of the reaction of o-xylene with BzCl over Hβ, the very low value (4.0 kcal mol−1) is evidence of a diffusional control.
| Reference | Reaction (parameter)a | EA/kcal mol−1 |
|---|---|---|
| a Kinetic coefficient (k1, k2) or rate (r) on which the evaluation of EA was based.b Range of values, according to the product isomer. | ||
| from Table 1 | ||
| 7 | benzene + BzCl over In2O3/Hβ (k1) | 17.6 |
| 9,10 | benzene + 1-dodecene over H3PW12O40/SiO2 (k2) | 10.9–11.5b |
| 11 | benzene + propene over CaHY (k2) | 23.6 |
| from Table 2 | ||
| 23 | anisole + BzCl over K10 (k1) | 15.6 ± 2.9 |
| benzene + BzCl over Clayzic (unactivated) (k1) | 14.2 ± 2.2 | |
| 26,27 | benzene + 1-dodecene over HY (k1) | 20.6 |
| 28 | benzene + 1-dodecene over HY (k1) | 12 ± 3 |
| 31 | benzene + BzCl over Ga2O3/HZSM-5 (k1) | 16.1 |
| 32 | benzene + BzCl over InCl3/K10 (k1) | 14.5 |
| benzene + BzCl over GaCl3/K10 (k1) | 15.1 | |
| benzene + BzCl over FeCl3/K10 (k1) | 11.6 | |
| from Table 3 | ||
| 36 | benzene + 1-dodecene over HY (r) | 15 |
| 41 | o-xylene + BzCl over Hβ (r) | 4.0 |
Table 5 shows the EA values available for additional reactions. In many cases they fall in the 10 to 20 kcal mol−1 range, but also values > 20 kcal mol−1 have been reported. In two cases values as low as 7–8 kcal mol−1 were measured, although the tests performed showed mass transfer resistances to be negligible.
| Reference | Reaction | EA/kcal mol−1 |
|---|---|---|
| a For these reactions tests are reported showing that mass transfer resistances were negligible. | ||
| 49 | phenol + 1-dodecene over Amberlyst-15a | 7.4 |
| 56 | benzene + propene over β zeolitea | 7.8 |
| 48 | phenol + isobutene over Amberlyst-15a | 12.3 (to p-alkylate) |
| 13.8 (to o-alkylate) | ||
| 46 | benzene + 1-dodecene over H4SiW12O40 | 13.8 |
| 52 | anisole + BzCl over Clayzic | 15.5 |
| 55 | benzene + propene over MCM-22 | 18.4 |
| 58 | phenol + methyl tert-butyl ether over H3PW12O40/K10a | 18.5 |
| 59 | benzene + 1-dodecene over H3PW12O40/K10a | 20.1 |
| 53 | p-cresol + isobutene over sulfated zirconiaa | 22.9 |
| 51 | p-cresol + isobutene over Amberlyst-15a | 23.2 |
| 54 | benzene + BzCl over sulfated zirconiaa | 28.0 |
Until now only the kinetic results for the mono-alkylation of aromatics have been considered; however, also kinetic data on consecutive dialkylation reactions are available. As shown in Table 6, the coefficient ratio for dialkylation and monoalkylation is in some cases < 1, in other cases ≅ 1, often > 1. The largest value was found during studies on the reaction of naphthalene with benzyl chloride over a MCM-41 catalyst: the second benzylation is reported as being much faster for 2-benzylnaphthalene than for the 1-benzyl isomer.15
| Reference | Reaction | T/°C | Ratio (dialkyl/monoalkyl) |
|---|---|---|---|
| 51 | p-cresol + isobutene over Amberlyst-15 | 65 | 0.21 |
| 56 | benzene + propene over β zeolite | 100 | 0.45 |
| 53 | p-cresol + isobutene over sulfated zirconia | 0.65 | |
| 46 | benzene + 1-dodecene over H4SiW12O40 | 65–150 | 1 |
| 6 | biphenyl + BzCl over HY | 80 | 1 |
| 11 | benzene + propene over CaHY | 230 | 1.15 |
| 12,13 | biphenyl + BzCl over amorphous silica-alumina | ||
| having Si/Al = 6.2 | 80 | 2 | |
| having Si/Al = 2.7 | 80 | 2.5 | |
| 13,14 | biphenyl + BzCl over HAlMCM-41 | 80 | 2.5 |
| 15 | naphthalene + BzCl over HAlMCM-41 | ||
| for 1-benzylnaphthalene | 80 | 1.4 | |
| for 2-benzylnaphthalene | 80 | 45 | |
| 16 | naphthalene + BzCl over Nafion–silica composite | 80 | 4.6 |
4 Catalyst stability
In several cases, the authors also investigated the possibility of re-using the catalyst one or more times after a (batch) run. Table 7 shows the average loss in activity per re-use, evaluated from tabulated data or plotted results.| Reference | Reaction | Catalyst | No. of re-uses | Average loss of activity per re-use |
|---|---|---|---|---|
| a when recycling is effected at reaction temperature | ||||
| 16 | naphthalene + BzOH | Nafion–silica composite | 3 | negligiblea |
| 31 | benzene + BzCl | Ga2O3/HZSM–5 | 3 | negligible |
| 32 | benzene + BzCl | InCl3/K10 | 5 | ∼1% |
| 7 | benzene + BzCl | In2O3/Hβ | 5 | ∼2% |
| 7 | benzene + BzCl | InCl3/Hβ | 5 | ∼5% |
| 54 | benzene + BzCl | sulfated ZrO2–Fe2O3 | 3 | ∼4% |
| 58 | phenol + MTBE | H3PW12O40/K10 | 3 | ∼5% |
| 59 | benzene + 1-dodecene | H3PW12O40/K10 | 2 | ∼6% |
| 8 | p-cresol + MTBE | sulfated ZrO2/K10 | 2 | ∼18% |
| 37 | o-xylene + styrene | Amberlyst–15 | 5 | ∼18% |
| 41 | o-xylene + BzCl | Hβ | 3 | ∼23% |
| 4 | benzene + BzCl | FeCl3/K10 or | ||
| FeCl3/SiMCM–41 | 1 | 40–50% | ||
| 54 | benzene + BzCl | sulfated ZrO2 | 1 | >87% |
A different way of testing catalyst stability is to use the catalyst for continuous runs over a period of at least several hours. Runs in the CSTR mode revealed amorphous low-alumina silicoaluminate (Si/Al = 6.2)12 and HAlMCM-4114 to have constant behaviour in the reaction of biphenyl with benzyl chloride; an analogous reaction using naphthalene again evidenced the stability of HAlMCM-4115 and, in the reaction of naphthalene with benzyl alcohol, stability was also found for the Nafion–silica composite.16 In contrast, the same kind of test showed a slight loss in activity for amorphous high-alumina silicoaluminate (Si/Al = 2.7).13 Runs in a fixed bed continuous reactor, for the reactions of benzene with ethene or propene, revealed MCM-22 to be stable.55
In other cases, the catalysts underwent regeneration and were then proved to be re-usable. This was the case of Hβ44 and Amberlyst-15.48
Among the reactions taken into account, some of the kinetic models of the alkylation processes included catalyst deactivation. In these cases, a deactivation rate coefficient kd was evaluated. The values reported in Tables 1–3 correspond to different kinetic equations, so their numerical values have to be interpreted. By integrating the kinetic equations, it can be seen that:
for HY (Si/Al = 7) in the benzylation of biphenyl,6 where kd = 0.25 min−1, this means that over 60 min the activity is lowered from 100% to about 6%;
for HAlMCM-41 in the same reaction,14 where kd < 0.001 min−1, this means that over 60 min the activity is lowered from 100% to a fraction > 94%;
for HY (Si/Al = 1.6) in the reaction of benzene with 1-dodecene,26,27 where kd = 0.31 min−1, this means that over 60 min the activity is lowered from 100% to 16%.
In the case of o-xylene reacting with styrene over Amberlyst-15,37kd = 0.056 min−1, the coke formation measured revealing that about 3% coke is deposited on the catalyst over 60 min.
5 Conclusions
The kinetic parameters for many monoalkylation reactions, over different types of solid acids, often at 40 to 100 °C, are reported in the scientific literature. In several cases, also apparent activation energy has been measured. In the present review, research papers including a discussion on resistances to mass transfer have been indicated.Aromatics like benzene, toluene, the xylenes, biphenyl, the phenols and anisole are the ones given the most consideration. The most common alkylating agents are benzyl chloride, olefins and alcohols. The most commonly employed catalysts are zeolites, supported metal chlorides and oxides, mesoporous (MCM) materials and heteropolyacids, these are followed by sulfated zirconia, ion-exchange resins, Nafion derivatives, and other minor items.
Data available on the kinetics of successive dialkylation reactions are also presented, the kinetic parameters usually being of the same order as those of the first reaction.
The catalysts were often tested for their stability, particularly keeping in mind their recycling for re-use: in several cases this was found possible, though usually with some loss of catalytic activity. Given the practical importance of catalyst re-use, further work is needed in this field.
References
- F. Arena, R. Dario and A. Parmaliana, Appl. Catal. A: General, 1998, 170, 127 CrossRef CAS.
- T. Okuhara, T. Nishimura, H. Watanabe and M. Misono, J. Mol. Catal., 1992, 74, 247 CrossRef CAS.
- T. Cseri, S. Békássy, F. Figueras and S. Rizner, J. Mol. Catal. A: Chemical, 1995, 98, 101 CrossRef CAS.
- V. R. Choudhary, S. K. Jana and A. S. Mamman, Microporous Mesoporous Mater., 2002, 56, 65 CrossRef CAS.
- P. H. J. Espeel, K. A. Vercruysse, M. Debaerdemaker and P. A. Jacobs, Stud. Surf. Sci. Catal., 1994, 84, 1457 CAS.
- P. Beltrame and G. Zuretti, Ind. Eng. Chem. Res., 1997, 36, 3427 CrossRef CAS.
- V. R. Choudhary, S. K. Jana, N. S. Patil and S. K. Bhargava, Microporous Mesoporous Mater., 2003, 57, 21 CrossRef CAS.
- G. D. Yadav, A. A. Pujari and A. V. Joshi, Green Chem., 1999, 1, 269 RSC.
- J Zhang, Z. Zhu, C. Li, L. Wen and E. Min, J. Mol. Catal. A: Chemical, 2003, 198, 359 CrossRef CAS.
- J. Zhang, B. Chen, C. Li, Z. Zhu, L. Wen and E. Min, Appl. Catal. A: General, 2003, 249, 27 CrossRef CAS.
- I. Iliuta, G. Bozga and M. Lupascu, Chem. Eng. Technol., 2001, 24, 933 CrossRef CAS.
- P. Beltrame and G. Zuretti, Ind. Eng. Chem. Res., 1998, 37, 3540 CrossRef CAS.
- P. Beltrame, G. Zuretti and F. Demartin, Ind. Eng. Chem. Res., 2000, 39, 1209 CrossRef CAS.
- P. Beltrame, F. Demartin and G. Zuretti, Appl. Catal. A: General, 2001, 218, 61 CrossRef CAS.
- P. Beltrame, F. Demartin and G. Zuretti, Appl. Catal. A: General, 2002, 232, 265 CrossRef CAS.
- P. Beltrame and G. Zuretti, Appl. Catal. A: General, 2003, 248, 75 CrossRef CAS.
- W. M. Meier, D. H. Olson, and Ch. Baerlocher, Atlas of zeolite structure types, 4th ed., in Zeolites, 1996, 17, special issue Search PubMed.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. K. Beck, Nature, 1992, 359, 710 CrossRef CAS.
- K. D. Schmitt, C. T.-W Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834 CrossRef CAS.
- P.-T. Tanev, M. Chibwe and T. J. Pinnavaia, Nature, 1994, 368, 321 CrossRef CAS.
- G. A. Olah, P. S. Iyer and G. K. Surya Prakash, Synthesis, 1986, 513 CrossRef CAS.
- M. A. Harmer, W. E. Farneth and Q. Sun, J. Am. Chem. Soc., 1996, 118, 7708 CrossRef CAS.
- S. J. Barlow, T. W. Bastock, J. H. Clark and S. R. Cullen, Tetrahedron Lett., 1993, 34, 3339 CrossRef CAS.
- S. J. Barlow, T. W. Bastock, J. H. Clark and S. R. Cullen, J. Chem. Soc., Perkin Trans. 2, 1994, 411 RSC.
- J. H. Clark, S. R. Cullen, S. J. Barlow and T. W. Bastock, J. Chem. Soc., Perkin Trans. 2, 1994, 1117 RSC.
- W. G. Liang, Z. Yu, Y. Jin, Z. Wang and Y. Wang, J. Chem. Technol. Biotechnol., 1995, 62, 98 CAS.
- W. G. Liang and J.-X. Zhu, Ind. Eng. Chem. Res., 1997, 36, 4651 CrossRef CAS.
- Y. Cao, R. Kessas, C. Naccache and Y. Ben Taarit, Appl. Catal. A: General, 1999, 184, 231 CrossRef CAS.
- H. H. P. Yiu, D. R. Brown and P. A. Barnes, Catal. Lett., 1999, 59, 207 CrossRef CAS.
- V. R. Choudhary and S. K. Jana, J. Catal., 2001, 201, 225 CrossRef CAS.
- V. R. Choudhary and S. K. Jana, Appl. Catal. A: General, 2002, 224, 51 CrossRef CAS.
- V. R. Choudhary and S. K. Jana, J. Mol. Catal. A: Chemical, 2002, 180, 267 CrossRef CAS.
- J. H. Clark, A. P. Kybett, D. J. Macquarrie, S. J. Barlow and P. Landon, J. Chem. Soc., Chem. Commun., 1989, 1353 RSC.
- P. S. Kumbhar, V. M. Yadav and G. D. Yadav, Chem. Modif. Surf., 1990, 3, 81 Search PubMed.
- B. Coq, V. Gourves and F. Figueras, Appl. Catal. A: General, 1993, 100, 69 CrossRef CAS.
- J. L. G. de Almeida, M. Dufaux, Y. Ben Taarit and C. Naccache, Appl. Catal. A: General, 1994, 114, 141 CrossRef.
- A. B. Dixit and G. D. Yadav, React. Funct. Polym., 1996, 31, 237 CrossRef CAS.
- T. Matsuda, T. Kimura, E. Herawati, C. T. Kobayashi and E. Kikuchi, Appl. Catal. A: General, 1996, 136, 19 CrossRef CAS.
- Q. Sun, M. A. Harmer and W. E. Farneth, Ind. Eng. Chem. Res., 1997, 36, 5541 CrossRef CAS.
- D. Bhattacharya, A. K. Pandey and A. P. Singh, Stud. Surf. Sci. Catal., 1998, 113, 737 CAS.
- A. P. Singh, B. Jacob and S. Sugunan, Appl. Catal. A: General, 1998, 174, 51 CrossRef CAS.
- B. Török, I. Kiricsi, A. Molnar and G. A. Olah, J. Catal., 2000, 193, 132 CrossRef CAS.
- M. A. Harmer, Q. Sun, A. J. Vega, W. E. Farneth, A. Heidekum and W. F. Hoelderich, Green Chem., 2000, 2, 7 RSC.
- X. C. Hu, G. K. Chuah and S. Jaenicke, Microporous Mesoporous Mater., 2002, 53, 153 CrossRef CAS.
- B. M. Lok, C. A. Messina, R. L. Patton, R. T. Gajek, T. R. Cannon and E. M. Flanigen, J. Am. Chem. Soc., 1984, 106, 6092 CrossRef CAS.
- R. T. Sebulsky and A. M. Henke, Ind. Eng. Chem., Proc. Des. Develop., 1971, 10, 272 Search PubMed.
- R. B. Wesley and B. C. Gates, J. Catal., 1974, 34, 288 CAS.
- P. N. Unni and S. Bhatia, J. Chem. Technol. Biotechnol., 1982, 33A, 1.
- H.-S. Park and S. K. Ihm, Korean J. Chem. Eng., 1985, 2, 69 Search PubMed.
- C. Buttersack, H. Widdecke and J. Klein, J. Mol. Catal., 1986, 38, 365 CrossRef CAS.
- E. Santacesaria, R. Silvani, P. Wilkinson and S. Carrà, Ind. Eng. Chem. Res., 1988, 27, 541 CrossRef CAS.
- A. Cornélis, P. Laszlo and S. Wang, Tetrahedron Lett., 1993, 35, 3849 CrossRef.
- G. D. Yadav and S. T. Tushar, Ind. Eng. Chem. Res., 1996, 35, 721 CrossRef CAS.
- S. N. Koyande, R. G. Jaiswal and R. V. Jayaram, Ind. Eng. Chem. Res., 1998, 37, 908 CrossRef CAS.
- A. Corma, V. Martinez-Soria and E. Schnoeveld, J. Catal., 2000, 192, 163 CrossRef CAS.
- M. Han, X. Li and S. Lin, Kinet. Katal., 2001, 42, 533 Search PubMed.
- G. D. Yadav, P. K. Goel and A. V. Joshi, Green Chem., 2001, 3, 92 RSC.
- G. D. Yadav and N. S. Doshi, Appl. Catal. A: General, 2002, 236, 129 CrossRef CAS.
- G. D. Yadav and N. S. Doshi, Org. Proc. Res. Dev., 2002, 6, 263 Search PubMed.
| This journal is © The Royal Society of Chemistry 2004 |