Polymer brushes via surface-initiated polymerizations
Steve
Edmondson
,
Vicky L.
Osborne
and
Wilhelm T. S.
Huck
*
Melville Laboratory for Polymer Synthesis, Department of Chemistry, Cambridge University, Lensfield Road, Cambridge, UK CB2 1EW. Tel: +44 1223 331509; Fax: +44 1223 334866; E-mail: wtsh2@cam.ac.uk
First published on 2nd December 2003
Abstract
Polymer brushes produced by controlled surface-initiated polymerization provide a route to surfaces coated with well-defined thin polymer films that are covalently bound to the substrate. All of the major controlled polymerization techniques have been applied to the synthesis of polymer brushes and examples of each are presented here. Many examples of brush synthesis in the literature have used the living atom transfer radical polymerization (ATRP) system, and in this tutorial review a particular focus is given to examples of this technique.
 Steve Edmondson Steve Edmondson | Steve Edmondson was born in Wirral, Merseyside in 1980. He received his Master’s degree from Cambridge University in 2002. He continued at Cambridge, and is currently in the first year of his PhD under Wilhelm Huck, investigating the synthesis and reactivity of functional polymer brushes. |
 Vicky L. Osborne Vicky L. Osborne | Vicky Osborne was born on the South East coast of Britain in 1974. She undertook her BSc at the University of Kent at Canterbury and is currently a final year PhD student at Cambridge University, a student of Wilhelm Huck. |
 Wilhelm T. S. Huck Wilhelm T. S. Huck | Wilhelm Huck was born in Sittard, The Netherlands in 1970. He received his MSc in chemistry (cum laude) from Leiden University in 1992, and did his PhD (1997) at Twente University working with Professor Reinhoudt on self-assembled metallodendrimers. After carrying out post-doctoral work with Professor Whitesides at Harvard, he took up his current position of ICI Lecturer at Cambridge University in 1999. His research interests include self-assembled monolayers, nanolithography and polymer brushes, and their applications in optoelectronic as well as biomedical devices. For his work on polymer brushes he was awarded the Young Researchers Medal 2001 of the RSC Macro Group UK. |
Introduction
The modification of surfaces with thin polymer films is widely used to tailor surface properties such as wettability, biocompatibility, corrosion resistance and friction. Such thin polymer films can be applied by depositing or spraying a polymeric coating from solution. Alternatively, polymers with reactive endgroups can be grafted onto surfaces, resulting in so-called polymer brushes. The advantage of polymer brushes over other surface modification methods (e.g. self-assembled monolayers) is their mechanical and chemical robustness, coupled with a high degree of synthetic flexibility towards the introduction of a variety of functional groups. There is also an increasing interest of using functional or diblock copolymer brushes for ‘smart’ or responsive surfaces, which can change a physical property (hydrophilicity, biocompatibility) upon an external trigger, such as heat (in the case of materials with a lower critical solution temperature, LCST), pH, or salt concentration (polyelectrolytes).Commonly, brushes are prepared by grafting polymers to surfaces, either via chemical bond formation between reactive groups on the surface and reactive endgroups, or by physisorption of block copolymers with ‘sticky’ segments. In this review, the emphasis will be on recent synthetic developments in preparing polymer brushes which broaden the scope of potential applications in which polymer brushes can play a pivotal role. The grafting to method is experimentally simple, but has some limitations. It is very difficult to achieve high grafting densities because of steric crowding of reactive surface sites by already adsorbed polymers. Furthermore, film thickness is limited by the molecular weights of the polymer in solution (films in the 100 nm thickness range are inaccessible). Relying on non-covalent adsorption of polymers to surfaces makes the adsorption a reversible process and such brushes are not stable under conditions where high shear forces are involved, for example. The introduction of functional groups might be hampered by the requirements for the physisorption to the surface (electrostatic or hydrophobic interactions), or the formation of a covalent bond via reactive groups on polymer and surface.
‘Surface-initiated polymerizations’ (also called grafting from) from initiators bound to surfaces are a powerful alternative to control the functionality, density and thickness of polymer brushes with almost molecular precision. First, the substrate of choice (planar or particle) is modified with initiator-bearing self-assembled monolayers. These monolayers can be formed on almost any surface, as long as the anchor functionality is chosen right (for example: thiols on gold, silanes on glass, Si/SiO2 and plasma oxidized polymers. The initiator surfaces are then exposed to solutions containing catalyst and monomer (plus solvent if necessary). Ideally, the polymerization is not only surface-initiated but also surface-confined, i.e. no polymerization in solution.
In order to achieve maximum control over brush density, polydispersity, and composition, plus at the same time allowing the formation of block copolymers on the surface, a controlled polymerization is highly desirable. Over the last few years, this field has rapidly evolved and all the major controlled polymerization strategies have been used to grow polymer brushes. Of these, controlled radical polymerizations (and most notably atom transfer radical polymerization, ATRP) have become the most popular routes, mostly because of their tolerance to a wide range of functional monomers and less stringent experimental conditions. Before reviewing radical polymerizations in more detail, an overview of the different controlled polymerizations that have been used for brush growth is presented.
Living ring opening polymerization
A number of commercially important polymers, such as polycaprolactone and polylactide, are produced by ring-opening polymerization (ROP). Thus, surface-initiated ROP is an attractive route to surfaces coated with thin layers of these polymers (Fig. 1).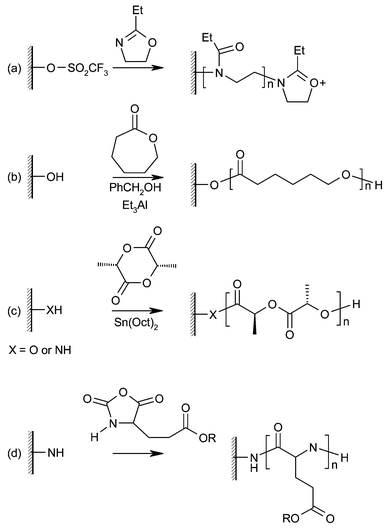 | ||
| Fig. 1 Polymer brushes grown by living ring opening polymerization (ROP). (a) Poly(N-propionylethyleneimine), (b) poly(ε-caprolactone), (c) poly(lactic acid), (d) poly(glutamate). | ||
Early work by Jordan and Ulman used the living cationic ROP of 2-ethyl-2-oxazoline to produce linear poly(N-propionylethyleneimine) (PPEI).1 A self-assembled monolayer (SAM) presenting trifluoromethane sulfonate (triflate) groups was prepared on a gold-coated glass slide by the adsorption of 11-hydroxyundecanethiol and subsequent vapour-phase functionalisation. Reaction with 2-ethyl-2-oxazoline in refluxing chloroform for seven days produced a 9 nm thick layer of PPEI (corresponding to a degree of polymerization of 10). Termination of this polymerization was achieved with the addition of N,N-dioctylamine, producing amphiphilic brushes. Although this reaction does not require the addition of a catalyst and produces well-defined brushes, the rate of propagation is extremely low compared to catalysed ROP or other controlled brush growth techniques. One possible explanation could lie in the instability of the initiator monolayer under the polymerization conditions.
Husseman et al. have used aluminium alkoxide catalysed ROP to grow brushes of poly(ε-caprolactone) (PCL) on gold surfaces.2 In contrast to the work of Jordan and Ulman, a SAM terminating in di(ethylene glycol) moieties was used to present OH groups for initiation. It was found that these SAMs gave more reproducible polymer growth and better long-term stability than simple long-chain alcohol SAMs. Organometallic catalysis with diethylaluminium alkoxides (prepared from triethylaluminium) allowed the formation of PCL brushes up to 70 nm thick at room temperature within a few hours. To achieve good control of brush thickness, it was necessary to add free initiator (benzyl alcohol) to the reaction mixture, the brush thickness being determined by the initial ratio of alcohol to ε-caprolactone. Exchange of the active site between bound and free polymer chains gives molecular weight control, but the polymerization is no longer surface-confined. Free polymer must be removed from the bound polymer brushes before analysis, so rigorous washing procedures must be employed. Interestingly, the soft-lithographic technique of micro-contact printing (μCP) was applied in this work to produce patterned brushes. Using an elastomeric stamp, a non-reactive thiol SAM was applied only to selected areas of the gold substrate. Immersion of the surface in a solution of initiator-functionalised thiol “backfilled” the rest of the surface with initiator. Brush growth now only occurred in the unprinted regions, giving a patterned surface representative of the stamp pattern. Brush height can then be determined directly using Atomic Force Microscopy (AFM).
Similar work by Choi and Langer explored the use of the tin(II) octoate catalyst to produce chiral poly(lactic acid) (PLA) brushes on gold and silicon substrates by ROP of L-lactide.3 PLA has attracted interest as a biodegradable polymer used in medical applications, and PLA brushes present a possible route to well-defined biocompatible surfaces with controlled release properties. Additionally, chiral brushes are of interest for coating surfaces for chiral separation and molecular recognition applications. Using an oligo(ethylene glycol) terminated SAM on gold, PLA brushes up to 12 nm thick could be grown in three days at 40 °C. Bulk and solution polymerization of L-lactide is usually carried out at higher temperatures, but the thermal instability of thiol SAMs on gold limits reaction temperatures. By moving to an amine SAM on a Si/SiO2 surface, which is stable at much higher temperatures, PLA brushes up to 70 nm thick were produced in three days at 80 °C. Notably, these polymerizations required no free initiator, i.e. they are surface-confined.
There have been several papers reporting the synthesis of polypeptide brushes with various side chains. For example, Schouten and co-workers. grew poly(L-glutamate) brushes from amine-functionalised silicon wafers and glass slides.4 The monomers used were N-carboxy anhydrides (NCA) of L-glutamates, which are cyclised amino acids that undergo ROP in the presence of amine groups. This polymerization is extremely versatile and allows the incorporation of a wide variety of side chains into the polymer brush. Brushes up to 40 nm thick were produced in only a few hours, and detailed study of the amide IR absorptions revealed the polypeptide brushes adopt an α-helix conformation on the surface. The “living” nature of the polymerization was demonstrated by the re-initiation of chain growth, and the synthesis of diblock copolymer brushes. Investigations of the electromechanical properties of a polyglutamate brush layer grown on an aluminium electrode have shown that the α-helical chain conformation adopted by the brushes confers piezoelectric properties.5
Living anionic polymerization
The highly living nature of anionic polymerization has made it an attractive choice for the synthesis of well defined polymer brushes (Fig. 2a,b). Jordan et al. used a SAM containing biphenyllithium groups to initiate the anionic polymerization of styrene on gold substrates.6 A SAM containing bromobiphenyl groups was formed initially, and converted to the initiating species by reaction with sec-butyllithium. On addition of styrene, very uniform films of 18 nm thickness were grown. The polymerization is slow, however, taking three days to reach this thickness.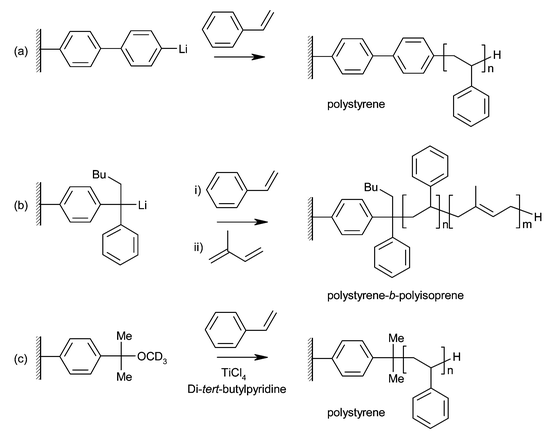 | ||
| Fig. 2 Polymer brushes grown by (a),(b) living anionic polymerization and (c) living cationic polymerization. | ||
In later work, Advincula et al. used SAMs containing diphenyethylene (DPE) groups, activated with n-butyllithium, to initiate the anionic polymerization of styrene.7 A thiol-containing SAM was used on gold substrates, while a dimethylchlorosilane SAM was used on silicon. Reaction times of several days again produced thin films (up to 16 nm), and results reported for silicon surfaces show large variations in film thickness for similar reaction conditions. The addition of tetramethylethylenediamine (TMEDA) additive produced a thicker film (26 nm). Block copolymer brushes of polystyrene-b-polyisoprene and polybutadiene-b-polystyrene were produced by the sequential addition of monomers, demonstrating the living nature of the reaction. Although living anionic polymerization is useful for the synthesis of well-defined brushes with low polydispersity, the extreme sensitivity of anionic polymerization to impurities necessitates the use of specialised glassware and rigorous purification and drying of reagents. This, coupled with restricted monomer functionality, long reaction times, and low values for final thickness of the polymer films, limits the use of this technique for polymer brush growth.
Living cationic polymerization
Little work has been done on the application of cationic polymerization to the synthesis of polymer brushes. In a study by Zhao and Brittain, SAMs terminating in cumyl methyl ether moieties were deposited on silicon wafer surfaces.8 Activation with TiCl4 in the presence of styrene and a proton scavenger (di-tert-butylpyridine) leads to the growth of brushes up to 30 nm thick in under an hour (Fig. 2c). Even with the addition of proton scavenger, initiation by protons leads to the formation of polystyrene in solution. Additionally, brush growth was carried out at −78 °C to suppress chain transfer reactions. The polymerization was shown to be living by re-initiation of the polymer chains to grow further polystyrene.Ring opening metathesis polymerization (ROMP)
Ring opening metathesis polymerization (ROMP) of strained cyclic monomers, in particular functionalised norbornenes, has attracted recent attention for the synthesis of polymers with useful electrical properties. Coating conductive substrates with well-defined brushes of these polymers may be of great use in the production of polymer electronic devices (Fig. 3).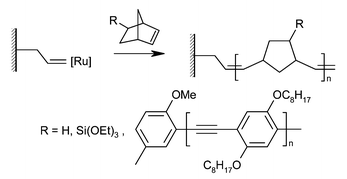 | ||
| Fig. 3 Polymer brushes grown by ring opening metathesis polymerization (ROMP) of norbornene-derived monomers. | ||
Whitesides and co-workers used an immobilised ruthenium catalyst to grow brushes from norbornene-derived monomers on silicon wafer surfaces.9 By forming a trichlorosilane SAM containing norbornene groups, and then exposing this to a solution of Grubbs-type ROMP catalyst, active surface-bound catalytic sites were produced. Exposure to solutions of norbornene-based monomers gave rapid but controlled brush growth, forming brushes up to 90 nm thick in 30 minutes. Exposure of these brushes to a solution of a second monomer allowed the growth of diblock copolymer brushes, characterised by transmission IR spectroscopy and an increase in the ellipsometric thickness of the brush layer. Microcontact printing (μCP) was also used to produce a patterned surface, with brushes only growing in well defined regions.
In similar work by Grubbs and co-workers, brushes were grown using norbornene as a monomer, again from silicon wafer surfaces.10 In an attempt to eliminate the electrically defective surface SiO2 layer normally present when brushes are grown from silicon, an alternative initiator attachment procedure was developed, allowing brushes with a direct Si–C bond to the surface to be synthesised. Some of the thickest films produced using surface-initiated polymerization (up to 5.5 µm) were grown in short times using a surface-bound ruthenium catalyst. Brushes anchored to the surface in this way provide a route to very well-defined insulating layers on silicon, with applications in electrical device construction. The internal structure of such brushes might no longer be reminiscent of typical brushes morphologies (e.g. stretched or collapsed) as it is inconceivable that one polymer chain stretches from the surface for several microns. In this case, re-initiation and chain transfer must occur to a certain degree.
More recently, Moon and Swager used ROMP to grow brushes with poly(p-phenylene ethynylene) “molecular wire” side chains for applications in chemical sensing.11 Immobilised ROMP catalyst groups were prepared using the same method as Kim et al. above, and used to polymerise a norbornene-capped poly(p-phenylene ethynylene) macromonomer. Brushes of up to 10 nm were produced, and re-initiation to form diblock copolymer brushes was achieved. In simple front-face fluorescence measurements, it was found that these brushes displayed several times brighter fluorescence than comparable spin-cast films, possibly due to reduced aggregation of chains.
Nitroxide-mediated polymerization (NMP)
Nitroxide-mediated polymerization (NMP), a living polymerization technique based on reversible capping of the active chain-end radical with a nitroxide leaving group, has been used to synthesise polymer brushes by Husseman et al. (Fig. 4a). Using surface-bound alkoxyamine initiator molecules on silicon wafers, polystyrene brushes over 100 nm thick were produced in 16 hours.12 Upon heating the initiator-functionalised wafer to 120 °C, the alkoxyamine moiety is cleaved off giving an alkyl radical and the stable nitroxide radical, TEMPO. Radical propagation is controlled by the reversible “capping” of the growing chain by the TEMPO radical, giving a living polymerization. This can be compared to atom transfer radical polymerization (ATRP), in which a halogen atom, transferred by a transition metal complex, mediates the propagation. Simply using surface-bound initiators alone did not give a controlled polymerization, however. The very small number of growing polymer chains, when compared to the monomer concentration, gives a very low overall concentration of free TEMPO and so inefficient capping of chain ends. The addition of “free” alkoxyamine initiator to the polymerization solution solves this problem, but leads to the formation of free polymer which must be removed from the brushes before analysis. The living nature of the polymerization was demonstrated by re-initiation of the capped chains to form block copolymer brushes.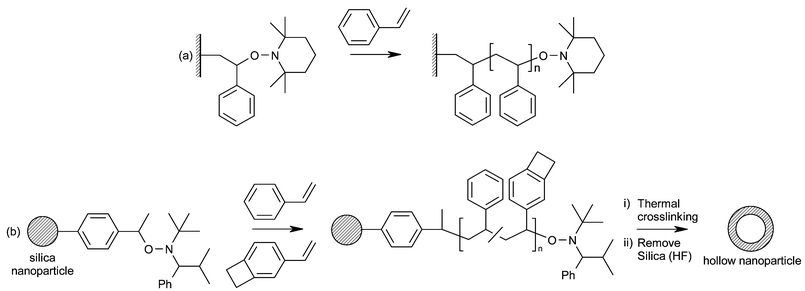 | ||
| Fig. 4 Polystyrene brushes and copolymer brushes grown by nitroxide-mediated polymerization (NMP). (a) Polystyrene, (b) random copolymer, leading to cross-linked hollow nanoparticles. | ||
In later work, Hawker and co-workers applied surface-initiated NMP to the formation of crosslinked, hollow nanoparticles, which may be of interest for applications such as drug delivery and as building blocks for nanotechnology (Fig. 4b).13 Functionalising silica nanoparticles with a trichlorosilane alkoxyamine initiator allowed the synthesis of polystyrene brushes, giving “core-shell” nanoparticles. Again, the addition of free alkoxyamine initiator is needed for good polymerization control. Extending this strategy, random copolymer brushes of styrene and a monomer which can be cross-linked were produced. Two monomers were tried: 4-vinylbenzocyclobutene, allowing thermal cross-linking at 220 °C, and maleic anhydride, which can be cross-linked with the addition of a diamine. The silica core of the nanoparticles was removed by treatment with aqueous hydrofluoric acid, giving hollow cross-linked polymer spheres.
Reversible addition–fragmentation chain transfer (RAFT) polymerization
RAFT polymerization is a controlled polymerization in which chain growth is initiated using a conventional technique (for example by a free radical initiator such as AIBN) and mediated by a dithioester chain transfer agent (Fig. 5). Radical transfer between growing chains, either those in solution or those on a surface, gives good control of the polymerization, and “capping” of growing chains by the dithioester moiety gives the reaction good living characteristics.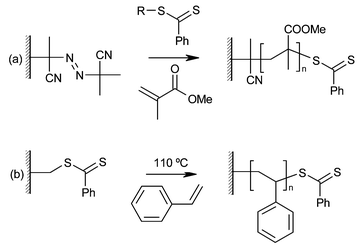 | ||
| Fig. 5 Polymer brushes grown by RAFT polymerization. (a) Poly(methyl methacrylate), (b) polystyrene. | ||
In a study by Baum and Brittain, a SAM containing an azo initiator group (like that in AIBN) was used to grow polymer brushes from silica surfaces in the presence of a dithiobenzoate chain transfer agent.14 At temperatures up to 90 °C and times of up to 48 hours, poly(methyl methacrylate) (PMMA) brushes with a thickness of 28 nm and poly(N,N-dimethylacrylamide) (PDMA) and polystyrene (PS) brushes up to 11 nm were grown. It was found that small amounts of untethered radical initiator (AIBN) were required in solution for brush growth to proceed. This was thought to be necessary to scavenge impurities which quickly terminate growing chains. However, this leads to the growth of polymer in solution, and brush samples must be extensively washed with dichloromethane before analysis. Although this polymerization is slow when compared to techniques such as ATRP or NMP, it is highly living. This was demonstrated by the re-initiation of the polymer chains to grow block copolymer brushes of PS-b-PDMA and PDMA-b-PMMA, and the sequential re-initiation of chains with the same monomer up to six times. A linear increase in brush thickness with re-initiation reaction number shows that virtually no termination reactions are occurring during brush growth.
In a study of the mechanism and kinetics of RAFT-mediated brush growth on silica particles, Tsujii et al. adopted a slightly different approach to that of Baum and Brittain. Using surface-initiated ATRP (see below), polystyrene brushes were grown from the silica particles for 1.5 hours at 110 °C in the presence of free ATRP initiator.15 The reversible “capping” of growing chain ends by a halogen atom allowed the polystyrene chains to be functionalised with a dithiobenzyl chain transfer agent after completion of the ATRP polymerization. This was achieved by heating the grafted chains, terminated with a bromine atom, with Cu(I)Br (to regenerate chain radicals), ligand and a dithiobenzoate chain transfer agent. Simply heating these functionalised chains with styrene at 110 °C, either with or without added “free” chain transfer agent, allowed RAFT-mediated brush growth to proceed for times of up to 24 hours. Radicals thermally produced in the bulk styrene attack the dithiobenzyl group on the grafted chains, and graft polymerization is initiated. In the case where free chain transfer agent is added, well controlled polymerization is achieved, with a linear increase of grafted chain molecular weight with conversion. Detailed study of the molecular weight distribution of the grafted chains, by dissolution of the silica particles by aqueous hydrofluoric acid and subsequent GPC analysis, reveals effective radical migration between these chains via successive chain transfer reactions. This helps to keep the polymerization well controlled, at the expense of a higher than expected rate of bimolecular termination between growing chains.
Atom transfer radical polymerization (ATRP)
Planar substrates
In recent years ATRP has been the most widely employed technique for the formation of polymer brushes via surface initiated polymerization. ATRP is compatible with a variety of functionalised monomers, and the living/ controlled character of the ATRP process yields polymers with a low polydispersity (MW/Mn) that are end functionalised and so can be used as macroinitiators for the formation of di- and triblock copolymers. Equally important, surface-initiated ATRP is experimentally more accessible than for example, the living anionic and cationic polymerizations discussed above, which require rigorously dry conditions. The synthesis of thiol and silane derivatised surface-bound initiators is easier than the AIBN–silane derivative or the nitroxide silane derivative for free radical and NMP polymerizations. The controlled nature of ATRP is due to the reversible activation–deactivation reaction between the growing polymer chain and a copper–ligand species.The most widely used monomer for the formation of polymer brushes via surface initiated ATRP is methyl methacrylate (MMA), the earliest example of which was published by Fukuda and co-workers in 1998.16 A monolayer bearing an ATRP initiator head group was immobilised onto silicon surfaces using the Langmuir–Blodgett (LB) technique to give a well-ordered array of initiating sites from which PMMA was grown. The addition of free initiator to the polymerization solution yields free polymer which can be characterised by conventional methods. The relatively narrow polydispersities of these polymers in conjunction with the fact that molecular weights were proportional to monomer conversion points towards the surface polymerization being controlled. PMMA film thicknesses of up to 80 nm in less than 12 hours were measured by ellipsometry. The thickness of the polymer brushes was related to the concentration of the free initiator added, the lower concentration of free initiator the thicker the films being achieved.
Matyjaszewski and co-workers reported controlled polymerizations without added free initiator;17 instead a CuII ligand complex was added to act as a deactivator. The addition of these complexes to the polymerization solution increased the initial CuII concentration to the same end as the addition of free initiator. Evidence of a controlled reaction was gained from a linear increase in ellipsometric thickness with time; identical yet separate experiments to form polymer in solution gave a graph showing a linear relationship between Mn and brush thickness. However, calculations of anticipated brush height for extended chains from polymer in solution showed that tethered polymer has a height of approximately one sixth of that calculated. Slower growth of the surface bound polymer due to geometric constraints resulting from high surface density of the chains could account for this observation. Alternatively, termination reactions from the dense initiator monolayer could play a role. Termination reactions throughout the polymerization would result in the loss of the terminal bromine atom from the chain thus preventing the re-initiation of the polymer brush to form block copolymer brushes. Polystyrene (PS) brushes (10 nm) were re-initiated to form PS-b-PMA block copolymer films, but reaction times of 20 hours were required to grow brushes with a total thickness of 100 nm. The length of time needed to achieve a PMA thickness of 90 nm points towards a much reduced initiator efficiency. This may be due to chain ends being buried in the polymer brush or termination reactions. PS-b-poly(tert-butyl acrylate) (PS-b-PTBA) brushes were synthesised and reflectance IR showed peaks corresponding to aromatic CH atoms and absorptions characteristic to esters of the PTBA. The PTBA block grown from PS films produced a decrease in the contact angle (90° to 86°). Hydrolysis of the PTBA layer to poly(acrylic acid) gave a sharp decrease in the contact angle to 18°.
The introduction of surface-initiated polymerizations in aqueous media saw the dawn of fast, controlled polymer brush growth at room temperature, even for water insoluble polymers such as PMMA and PGMA. Surface polymerization at lower temperatures has several advantages; firstly these polymerizations are compatible with substrates that are sensitive to elevated temperatures, for example, thiol SAMs on gold. In addition, spontaneous thermal polymerization and other reactions such as transesterification, elimination reactions and thermal cross-linking are less likely. Jones and Huck18 reported the formation of 30 nm thick PMMA brushes grown in aqueous media in only 35 min, which compares favourably to previously reported reaction times of up to 12 hours. The controlled nature of the polymerization was demonstrated by the linear time vs. thickness plot and by the synthesis of PMMA-b-PHEMA films. PGMA and PHEMA brushes were also grown with the PGMA films reaching a thickness of 125 nm in 90 mins and retaining the epoxy groups, which have potential for further derivatisation.
Water accelerated ATRP was shown to be able to grow very thick polymer films in short reaction times. Huang et al. used surface initiated ATRP from SAMs on Au to grow 700 nm thick PHEMA films in just 12 hours.19 Control experiments of polymerization solutions consisting of neat monomer, catalyst and ligand gave polymer films of 6 nm in an equivalent time, thus demonstrating the accelerating effect of water on surface initiated ATRP. The ability to extend the films with poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) confirmed the presence of dormant sites at the termini of PHEMA chains. The PDMAEMA film thickness when grown from a PHEMA brush was comparable to that when the PDMAEMA homopolymer was initiated directly from an initiator SAM. It can therefore be assumed that most grafted polymer chains retained their end functionality. The hydroxy groups of PHEMA can serve as reactive sites with a variety of functional groups. Bruening and co-workers reacted the PHEMA films with acetyl chloride and cinnamoyl chloride. These reactions were almost quantitative as indicated by the disappearance of the hydroxy peak (3500–3300cm−1) in reflectance FTIR spectra and a strong increase in the intensity of the carbonyl peak for esters. The PHEMA brushes also underwent an increase in thickness upon derivatisation in line with the increased volume of the repeat unit.
Room temperature ATRP was utilised by Bruening and co-workers to grow cross-linked films of ethylene glycol dimethacrylate (EGDMA).20 These brushes have pendant methacrylate groups which lead to cross-linked polymer films that have better mechanical and chemical stability than linear polymer brush analogues (Fig. 6). A surface confined polymerization is essential because the formation of polymer in solution will result in gelation or precipitation creating an inhomogeneous film. The polymerization system was similar to that of the Huck group, but with a water/DMF solvent (in place of water/methanol) being used to produce a homogenous system when using water-immiscible monomers. The living character of the polymerization was demonstrated by the re-initiation of the polymer films to give an increase in thickness. Swelling of the cross-linked films in THF gave results consistent with a highly cross-linked polymer network.
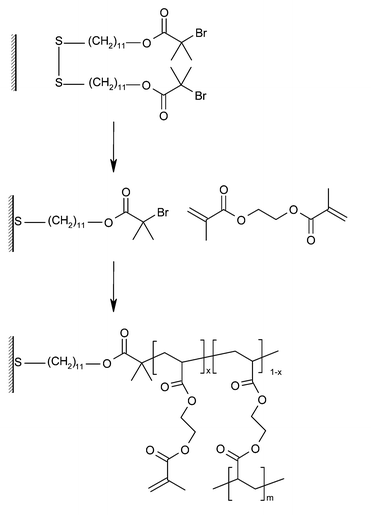 | ||
| Fig. 6 Cross-linked polymer brushes grown by ATRP of ethylene glycol dimethacrylate. | ||
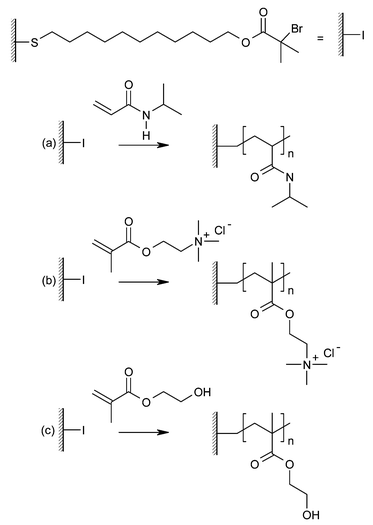 | ||
| Fig. 7 Polymer brushes grown by ATRP. (a) Poly(N-isopropylacrylamide) (PNIPAM), (b) poly(2-(methacyloyloxy)ethyl trimethylammonium chloride) (PMETAC), (c) poly(hydroxyethyl methacrylate) (PHEMA). | ||
In later work, Bruening et al.21 used a similar procedure to produce PDMAEMA brushes and a purely aqueous polymerization to produce PHEMA brushes. To demonstrate the living nature of these polymerizations block copolymers of PHEMA-b-PDMAEMA were also formed. To ensure high retention of end-group functionality for the initiation of the second block, the polymerization of the PHEMA block was quenched with a solution of CuBr2/bpy. Interestingly, this paper also studies the effect of mixed halide catalyst/deactivator systems. It was found that CuBr/CuBr2 did not afford sufficient control, whereas CuCl/CuBr2 systems gave a more linear increase in brush thickness with time, indicating a more ‘living’ character. It is proposed that this is due to the higher strength of the C–Cl bond than the C–Br bond.
The effectiveness of quenching with CuBr2/bpy was further investigated by the formation of triblock copolymer brushes. Quenching of a polymer brush with CuBr2/ligand solution effectively stops polymerization whilst promoting the retention of the Br atom at the chain ends and promoting the suitability of brushes for the subsequent polymerization of sequential polymer blocks. Using a quenching and re-initiation (QR) approach Bruening and co-workers grew PMA-b-PMMA-b-PHEMA brushes on Au, as evidenced using ellipsometry and reflectance FTIR. The efficiency of the QR scheme was confirmed via the synthesis of triblock PMMA, a homopolymer formed through three subsequent QR cycles. The PMMA polymer was detached from the Au surface using iodine and the molecular weight distribution determined using GPC. The Mn and increased PDI of the triblock PMMA compared to the single initiation PMMA points towards a small degree of the termination during the QR cycles. The efficiency of the QR scheme was also compared with a simple washing procedure. Heptablock PMMA films were prepared and as the number of QR cycles increased the difference in the thickness of the films prepared by the two methods also increased, indicating that some active chains terminate during solvent washing, resulting in fewer chains growing and hence lower thicknesses.
The Huck group have applied aqueous ATRP to the production of poly (N-isopropylacrylamide) (PNIPAM) on gold surfaces.22 Brushes of this type show temperature dependant hydrophilic/hydrophobic properties and are possible candidates for smart surfaces. The properties of these patterned brushes were probed using AFM under water. It was shown that reversible changes in topography, morphology, stiffness and adhesion occurred, consistent with a phase transition.
Surface ATRP has been shown to be compatible with other forms of polymerization. Brittain and co-workers23 synthesized PS-b-PMA and PS-b-PMMA brushes via sequential carbocationic polymerization and ATRP from initiator terminated SAMs. In this instance diblock brushes were of interest because of the phase separation properties of diblock copolymers. The behaviour of these tethered diblock brushes, as explored by AFM, revealed the formation of various nanomorphologies which was dictated by the chemical and physical composition of the brush and the solvent it was exposed to (Fig. 8). A brush consisting of PS-b-PMMA showed a smooth featureless surface after treatment with DCM and contact angles in good agreement with previously reported values for PMMA thin films. The same brush exhibited advancing contact angles close to that reported for PS after immersion in cyclohexane. The AFM revealed an irregular wormlike structure and increased surface roughness. It is speculated that the PS blocks are swollen by the cyclohexane, whilst the PMMA blocks migrate to avoid contact with the solvent. This structure is more pronounced in films composed of a thinner PMMA layer, where a higher mobility is available to shorter chains. Re-exposure of these films to DCM resulted in featureless films with PMMA advancing and receding contact angles, showing that the rearrangement of the films is reversible.
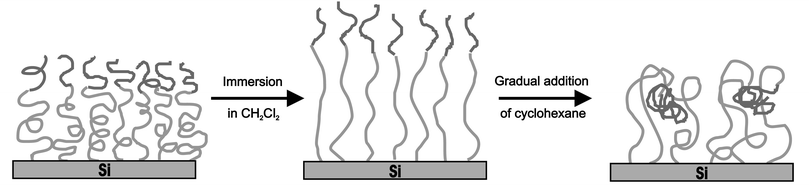 | ||
| Fig. 8 Surface morphology changes of PS-b-PMMA diblock copolymer brushes upon treatment with different solvents. | ||
Tethered PS-b-PMA brushes showed a different morphology to the PS-b-PMMA films on exposure to DCM. Rather than revealing a smooth featureless surface, a rough interconnected network with numerous isolated pits was observed by AFM. Solvent mixtures of DCM and cyclohexane at different ratios yield surfaces with varying degrees of roughness. The seemingly periodic nanomorphology is caused by the soluble block forming a layer around the insoluble core, resulting in an array of surface-immobilized micelles. The length of each polymer block determines the size of the domains and the surface roughness, however the overall morphology of the surface is the same. ABA type triblock copolymer brushes of PS and PMA showed similar reversible morphologies24 as the diblocks.
Surface-initiated ATRP has also been exploited to grow polyelectrolyte brushes as shown by Huck and co-workers.25 Both cationic and anionic brushes were synthesised in a controlled fashion using charged monomers, thus ensuring a charge is carried on every monomer and dispensing with the need for a post-polymerization charge introduction step. The controlled nature of the polymerizations was illustrated by the growth of triblock copolymer brushes consisting of cationic and anionic polymers separated by a neutral moiety, poly(2-(methacryloyloxy)ethyl trimethylammonium chloride)-b-PMMA-b-poly(sodium methacrylate) (PMETAC-b-PMMA-b-PNaMA).
An alternative route to polyelectrolyte brushes was published by Boyes et al.26 which involved the controlled polymerization of tert-butyl acrylate which was subsequently hydrolysed by aqueous HCl to yield poly(acrylic acid) brushes. These brushes can then be further derivatised to the silver salt by reaction with silver acetate solution. The formation of polyelectrolyte brushes and PS-b-PAA(Ag+) block copolymer brushes was followed using ellipsometry, contact angle goniometry and FTIR. Contact angles were employed to follow the switching of the brushes on exposure to different solvents.
Various theoretical and experimental approaches to polymer brushes have revealed that the grafting density (σ) of the polymers dictates whether a ‘brush’ or ‘mushroom’ regime is adopted. The issue of initiator density was addressed by Genzer and co-workers.27 A gradient density initiator SAM on silicon substrates was formed, with any spaces subsequently back filled with methyl terminated trichlorosilane to minimise any physisorption of monomer and/or polymer formed in solution on the areas of the substrate not containing the initiator SAM. Polyacrylamide (PAAm) was grown from the surface using ATRP and ellipsometry was used to measure the thickness of the polymer film as a function of the position on the substrate. It was found that the thickness of the film decreased gradually with decreasing initiator density. Plotting the polymer thickness against PAAm grafting density on the substrate showed that at low σ height is independent of grafting density, indicating that the polymer chains are in the ‘mushroom’ regime. Conversely, at higher polymer grafting densities, height of the polymer increases with increasing grafting density symptomatic of a polymer brush. This study found that the crossover between the ‘mushroom’ and ‘brush’ regimes occurred at σ ≈ 0.0065 nm−2.
The effect of initiator density on polymer brush growth by ATRP was also studied by Huck and coworkers.28 By forming a SAM on gold-coated mica from a mixture of α-bromoester initiator thiol and inert undecane thiol, a mixed monolayer with a controlled dilution of initiating groups was produced. An almost linear increase in thickness with initiator concentration, from 0 to 100%, was observed for PMMA brushes grown for the same time. At lower initiator concentrations, the growing chains have more room to spread laterally across the surface, producing an apparently lower thickness of the polymer layer. Interestingly, the brushes did not show a maximum initiator density above which no further thickness increase is observed. By cleaving the polymers from a large area of gold-coated wafer, and subsequent analysis by GPC, the number average molecular weight of the PMMA brushes could be determined. From this value, a typical cross-sectional area of a brush chain could be estimated, and was found to be ∼170 Å2 for a 28 nm brush on a 100% initiator SAM. Since the surface area of one initiator molecule is ∼20 Å2, it would be expected that each polymer brush chain would cover 10–12 initiator molecules. If this was true, brush thicknesses on SAMs between 10% and 100% initiator concentration should be similar, which is not the case. This discrepancy could possibly be explained by poor initiator efficiency at the start of the reaction.
Colloidal substrates
Surface-initiated ATRP has not been confined to planar surfaces; Huang and Wirth extended the procedure to colloidal supports.29 Polyacrylamide was grown from porous silica gel and shown to be suitable for the separation of proteins by size exclusion. It was calculated that the thickness of the film was significantly less than the average pore diameter of the silica so that the polyacrylamide film would reduce the size of the pores but not block them. Separation of a mixture of four proteins was performed at neutral pH, where the charge on silica is nearly maximized. The absence of strong electrostatic interactions indicated a homogenous covering of the silica by the polymer film.The effect of polymers grafted from silica supports on the flocculation of the particles was investigated by Armes and co-workers.30 300 nm Stöber silica was coated with a silane ATRP initiator from which various hydrophilic monomers were polymerized (Fig. 9). Brushes produced included poly(oligo(ethylene glycol)methacrylate) (POEGMA) and poly(2-(N-morpholino)-ethyl methacrylate) (PMEMA). PMEMA exhibits LCST at approximately 34 °C and the PMEMA–silica particles began aggregating at this temperature, as shown by light-scattering measurements. Complete re-dispersion of the flocculated silica particles occurred on cooling. The aqueous solution properties of the grafted polymer chains determine the colloidal stability of the particles.
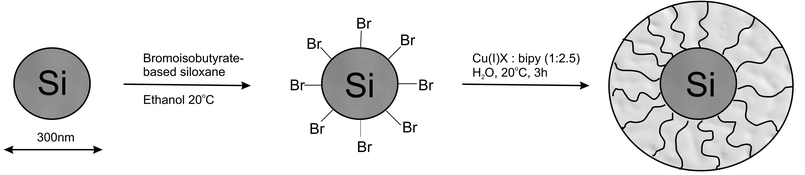 | ||
| Fig. 9 Polymer brushes grown from silica nanoparticles. | ||
The colloidal properties of polymer coated silica was further investigated with polyelectrolyte grafted silica particles.31 The polyelectrolytes included were poly(sodium 4-styrenesulfonate) (PSStNa), poly(sodium 4-vinylbenzoate (PNaVBA), PDMAEMA and poly(2-(diethylamino)ethyl methacrylate) (PDEA). These polymerizations were at ambient temperature (except PDEA at 50 °C) in protic media. The polyelectrolyte brushes enhanced the stability of the colloidal particles, preventing flocculation in aqueous media due to the electrostatic repulsive forces. As expected, the weak polyelectrolyte-covered silica particles flocculated at the pH where the brushes become neutral, the strong polyelectrolyte brush–silica particle is soluble at all pH ranges above the isoelectric point. The colloidal stability of the polyelectrolyte-grafted silica particles is dictated by the qualities of the polyelectrolyte brush.
Polymeric substrates
Of recent interest is the application of surface initiated polymerization to polymeric substrates rather than inorganic surfaces. To date, there have been several studies of surface initiated polymerization from polymer microspheres, but little work has been carried out using macro-scale polymer surfaces.Wu, Efimenko and Genzer have used a technique they have called mechanically assisted polymer assembly (MAPA) to produce polymer brushes of polyacrylamide on a cross-linked polydimethylsiloxane (PDMS) surface.32 The MAPA technique involves first stretching the PDMS substrate, then generating silanol (Si–OH) groups on the surface by exposure to UV/O3. A trichlorosilane ATRP initiator was then deposited onto the surface from the vapour phase, where reaction with the surface silanol groups produces tethered initiator. Polyacrylamide brushes were then grown under ATRP conditions at 130 °C. The strain was then released, allowing the PDMS substrate to return to its former size, producing densely grafted polymer brushes. By altering the amount by which the PDMS substrate is stretched, the brush grafting density can be controlled.
Bontempo et al.33 have grown a variety of brushes from polystyrene microspheres using aqueous ATRP, including PNIPAM, PHEMA, poly(poly(ethylene glycol)-1100 monomethacrylate) and block copolymers thereof. It was found that by varying the conditions of the polymerization, it was possible to induce brush growth either only on the surface of the microsphere, or throughout its bulk. Interestingly, it was found that, after functionalisation of the surface to act as an ATRP initiator, pure polystyrene microspheres would not initiate polymerization of hydrophilic monomers. Initiation only occurred if either the polystyrene microspheres also contained poly(ethylene glycol), or were ‘primed’ by first growing poly(poly(ethylene glycol)–1100 monomethacrylate) brushes.
Other work using polymer microspheres to initiate ATRP includes that by Zheng and Stöver, who grew PMA, PMMA, PHEMA, PDMAEMA and block copolymer brushes from functionalised divinylbenzene/hydroxyethyl methacrylate copolymer microspheres at room temperature.34 Polymer grafted spheres were characterized using ESEM, FTIR, Coulter particle sizing and potentiometric titration. These polymer grafted microspheres have potential applications in the binding and separation of proteins and other biomolecules.
Guerrini et al. used cross-linked poly[styrene-co-2-(2-bromopropionyloxy)] latex particles to initiate the ATRP of 2-hydroxyethyl acrylate (HEA) and 2-(methacryloyloxy)ethyl trimethylammonium chloride (METAC), giving particles with a hydrophobic core and a hydrophilic shell.35 Finally, Jayachandran et al. used polystyrene latex particles to initiate the aqueous ATRP of N,N-dimethylacrylamide (DMA).36 A variety of catalyst systems were investigated in an effort to produce well controlled polymerizations.
Conclusions
In conclusion, the surface-initiated route to polymer brushes has tremendously expanded the application range of polymer brushes. It is now possible to grow brushes on virtually every surface, to any thickness, of every composition, incorporating a multitude of functional groups and containing series of blocks. All modern polymer synthetic strategies can be extended towards polymer brushes. This in itself has been a very rewarding path of discovery over the last 5 years and will certainly continue to be so for some time. At the same time, it also opens up new possibilities for creating ‘smart’ or responsive surfaces, and we have only seen the beginning of research in this direction. Polymer brushes have interesting physical properties that are primarily related to the fact that the polymers are covalently tethered to the surface while the other end of the chain is freely moving in solution. No doubt, this will lead to new applications that are only possible because of the unique brush properties. Considering that surface modifications through coatings with polymer films are currently being used in myriad applications, we envisage that polymer brushes will become a completely new solution to many problems involving interfaces.References
- R. Jordan and A. Ulman, J. Am. Chem. Soc., 1998, 120, 243–247 CrossRef CAS.
- M. Husseman, D. Mecerreyes, C. J. Hawker, J. L. Hedrick, R. Shah and N. L. Abbott, Angew. Chem. Int. Ed., 1999, 38, 647–649 CrossRef.
- I. S. Choi and R. Langer, Macromolecules, 2001, 34, 5361–5363 CrossRef CAS.
- R. H. Wieringa, E. A. Siesling, P. F. M. Geurts, P. J. Werkman, E. J. Vorenkamp, V. Erb, M. Stamm and A. J. Schouten, Langmuir, 2001, 17, 6477–6484 CrossRef CAS.
- T. Jaworek, D. Neher, G. Wegner, R. H. Wieringa and A. J. Schouten, Science, 1998, 279, 57–60 CrossRef CAS.
- R. Jordan, A. Ulman, M. H. Rafailovick and J. Sokolov, J. Am. Chem. Soc., 1999, 121, 1016–1022 CrossRef CAS.
- R. Advincula, Q. G. Zhou, M. Park, S. G. Wang, J. Mays, G. Sakellariou, S. Pispas and N. Hadjichristidis, Langmuir, 2002, 18, 8672–8684 CrossRef CAS.
- B. Zhao and W. J. Brittain, Macromolecules, 2000, 33, 342–348 CrossRef CAS.
- N. Y. Kim, N. L. Jeon, I. S. Choi, S. Takami, Y. Harada, K. R. Finnie, G. S. Girolami, R. G. Nuzzo, G. M. Whitesides and P. E. Laibinis, Macromolecules, 2000, 33, 2793–2795 CrossRef CAS.
- A. Juang, O. A. Scherman, R. H. Grubbs and N. S. Lewis, Langmuir, 2001, 17, 1321–1323 CrossRef.
- J. H. Moon and T. M. Swager, Macromolecules, 2002, 35, 6086–6089 CrossRef CAS.
- M. Husseman, E. E. Malmström, M. McNamara, M. Mate, D. Mecerreyes, D. G. Benoit, J. L. Hedrick, P. Mansky, E. Huang, T. P. Russell and C. J. Hawker, Macromolecules, 1999, 32, 1424–1431 CrossRef.
- S. Blomberg, S. Ostberg, E. Harth, A. W. Bosman, B. Van Horn and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1309–1320 CrossRef CAS.
- M. Baum and W. J. Brittain, Macromolecules, 2002, 35, 610–615 CrossRef CAS.
- Y. Tsujii, M. Ejaz, K. Sato, A. Goto and T. Fukuda, Macromolecules, 2001, 34, 8872–8878 CrossRef CAS.
- M. Ejaz, S. Yamamoto, K. Ohno, Y. Tsujii and T. Fukuda, Macromolecules, 1998, 31, 5934–5936 CrossRef CAS.
- K. Matyjaszewski and P. J. Miller, Macromolecules, 1999, 32, 8716–8724 CrossRef CAS.
- D. M. Jones and W. T. S. Huck, Adv. Mater., 2001, 13, 1256–1259 CrossRef CAS.
- W. Huang, J.-B. Kim, M. L. Bruening and G. L. Baker, Macromolecules, 2002, 35, 1175–1179 CrossRef CAS.
- W. Huang, G. L. Baker and M. L. Bruening, Angew. Chem. Int. Ed., 2001, 40, 1510–1512 CrossRef CAS.
- J. B. Kim, W. X. Huang, M. L. Bruening and G. L. Baker, Macromolecules, 2002, 35, 5410–5416 CrossRef CAS.
- D. M. Jones, J. R. Smith, W. T. S. Huck and C. Alexander, Adv. Mater., 2002, 14, 1130–1134 CrossRef CAS.
- B. Zhao, W. J. Brittain, W. Zhou and S. Z. D. Cheng, Macromolecules, 2000, 33, 8821–8827 CrossRef CAS.
- S. G. Boyes, W. J. Brittain, X. Weng and S. Z. D. Cheng, Macromolecules, 2002, 35, 4960–4967 CrossRef CAS.
- V. L. Osborne, D. M. Jones and W. T. S. Huck, Chem. Commun., 2002, 1838–1839 RSC.
- S. G. Boyes, B. K. Mirous and W. J. Brittain, Polym. Prepr., 2003, 44, 552–553 Search PubMed.
- T. Wu, K. Efimenko and J. Genzer, J. Am. Chem. Soc., 2002, 124, 9394–9395 CrossRef CAS.
- D. M. Jones, A. A. Brown and W. T. S. Huck, Langmuir, 2002, 18, 1265–1269 CrossRef CAS.
- X. Huang and M. J. Wirth, Anal. Chem., 1997, 69, 4577–4580 CrossRef CAS.
- C. Perruchot, M. A. Khan, A. Kamitsi, S. P. Armes, T. von Werne and T. E. Patten, Langmuir, 2001, 17, 4479–4481 CrossRef CAS.
- X. Y. Chen, D. P. Randall, C. Perruchot, J. F. Watts, T. E. Patten, T. von Werne and S. P. Armes, J. Colloid Interface Sci., 2003, 257, 56–64 CrossRef CAS.
- T. Wu, K. Efimenko, P. Vlcek, V. Subr and J. Genzer, Macromolecules, 2003, 36, 2448–2453 CrossRef CAS.
- D. Bontempo, N. Tirelli, K. Feldman, G. Masci, V. Crescenzi and J. A. Hubbell, Adv. Mater., 2002, 14, 1239–1241 CrossRef CAS.
- G. Zheng and H. D. H. Stöver, Macromolecules, 2002, 35, 7612–7619 CrossRef CAS.
- M. M. Guerrini, B. Charleux and J.-P. Vairon, Macromol. Rapid Commun., 2000, 21, 669–674 CrossRef CAS.
- K. N. Jayachandran, A. Takacs-Cox and D. E. Brooks, Macromolecules, 2002, 35, 4247–4257 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2004 |