Sports drug testing – an analyst's perspective
Graham J.
Trout
and
Rymantas
Kazlauskas
Australian Sports Drug Testing Laboratory, Australian Government Analytical Laboratories, Sydney, Australia. E-mail: graham.trout@agal.gov.au; Fax: 61 2 9449 8080; Tel: 61 2 94490111
First published on 8th December 2003
Abstract
Sport plays a major role in the lives of many people, both for active participation and as entertainment. Sport is now a huge nationally and internationally based industry. The desire to win has led some athletes to resort to the use of performance enhancing drugs. With huge financial rewards now available in some sports the pressure to excel has grown. Some have argued that drug use should be given free rein, however most people are of the view that it is athletic prowess that should be applauded not the efficacy of various performance enhancing drugs. Apart from the obvious aspects of equality and fair play, the use of drugs is associated with significant health risks. In the 1960's the use of stimulants in sports such as cycling led to the death of at least one cyclist. Since 1968 the International Olympic Committee (IOC) has required all Olympic Games’ host cities to provide laboratory facilities for the analysis and detection of performance enhancing drugs. There are now 29 IOC accredited laboratories throughout the world that routinely test samples from athletes for the presence of such drugs. The purpose of this tutorial review is to give an overview of drug testing procedures, including those that were used at the last summer Olympic Games in Sydney 2000, and the incorporation of the latest developments in analytical chemistry technology in the drug testing process. More recently, developments in biotechnology mean that the use of whole new classes of drugs are banned in sport, often requiring new methodologies and techniques for their analysis. The contest between those who wish to cheat and those who wish to maintain fair play in sport is an ongoing one.
 Graham J. Trout Graham J. Trout | Graham Trout obtained a PhD from the University of Sydney in 1973. After periods in industrial research and teaching he joined the Australian Government Analytical Laboratories in 1988. Since then he has been involved in various degrees with the development of sports drug testing. He is currently the deputy director of the ASDTL with the primary responsibility of developing new methods. Career highlights have included drug testing at the Sydney Olympics with the introduction of a test for erythropoietin, and a feature article in Sports Illustrated. |
 Rymantas Kazlauskas Rymantas Kazlauskas | Rymantas Kazlauskas obtained a PhD from the University of Sydney in 1972. He worked in a post doctoral position in Cambridge (UK) then spent 8 years enjoying chemistry of marine natural products at Roche in Sydney. After a brief stay at the Pharmacology department at University of Sydney he joined the Australian Government Analytical Laboratories in 1986. Since 1988 he has been the Director of the sports drug testing facility (ASDTL) which tests most athletes in the Australasian region with the occasional increase in sample load for events such as an Olympic Games. |
1 Introduction
The use of drugs to enhance performance has been a problem for sport for many years. There is some evidence that elixirs were used in ancient times in an attempt to increase athletic prowess. However in modern times, the systematic testing of athletes for performance enhancing drugs did not begin until 1968 with testing for stimulants such as amphetamine.1 A comprehensive review of advances in the detection of performance enhancing drugs was published in 1997.2,3 This review includes recent developments in doping control.The list of banned substances prepared by the IOC and the World Anti-Doping Agency (WADA) describes those substances that may be used to provide an unfair advantage for an athlete. The IOC in their 2002 anti-doping code listed five classes of prohibited substances as well as prohibited methods. The classes listed were stimulants, narcotics, anabolic agents, diuretics, and ‘peptide hormones, mimetics and analogues’. The IOC/WADA 2003 code4 has been expanded to seven classes of prohibited substances with the new classes being ‘agents with ‘an anti-oestrogenic activity’ and ‘masking agents’. A summary of the banned list is shown in Fig. 1. The prohibited methods include ‘enhancement of oxygen transfer’, ‘pharmacological, chemical and physical manipulation’, and ‘gene doping’. Examples are given within each class of substances but the list provided is not intended to be drug exhaustive and the phrase “and related substances” is included in each class. In addition some classes of compounds such as beta-blockers are prohibited only for use during competition in specific sports. The classes largely group compounds in terms of their pharmacological effects and often drugs with similar pharmacological properties have similar chemistry.
 | ||
| Fig. 1 The 2002/3 list of substances banned in sport. | ||
2 Detection of banned drugs
The first stage in the detection of banned substances is the collection of the sample for analysis. The laboratory plays no direct part in this process, which allows the maintenance of athlete anonymity. The quality of such sampling and subsequent sample handling is critical to the entire process since it is necessary to be able to unambiguously link the result from an analysis back to the athlete who produced the sample. The IOC has had detailed procedures as part of their anti-doping code for several years and since 1999 an international standard has been available.5 Special security kits are used for sample collection with uniquely labelled glass bottles and caps. A split sample is sent to the laboratory and the bottles are separately labelled the A sample and the B sample. The samples have a set of accompanying chain of custody documents that allow all stages of the sample movement to be monitored. At the laboratory the A sample is analysed and results reported. The intact B sample is retained frozen until after a positive result is detected in the A sample, upon which time the athlete has the right to ask for it to be opened in front of witnesses, who may also attend the subsequent analysis procedure. These processes are subject to review as part of the laboratory's mandatory ISO17025 accreditation.The detection of drug use is not a trivial analytical task. Fig. 2 outlines the many questions to be answered before the actual analysis can begin. What is the best material to collect for analysis? The obvious choices are blood and urine although saliva and hair have been proposed. For most drugs, urine is the best choice for a number of reasons. Its collection is non-invasive, the volume available is fairly large, and perhaps most importantly the concentration of most of the drugs to be detected is higher in urine than in blood. However for some peptide hormones (e.g. growth hormone) blood will be the matrix of choice. Having decided on the substrate to be analysed, the next question is what analytes are to be measured. For some drugs a significant proportion of the parent compound is excreted unchanged, whilst others are changed extensively by the body's metabolism. Thus knowledge of biological processes is required to assist the identification of metabolites that may be present. For drugs that have routine medical applications this information is readily available in the literature. However athletes do not restrict themselves to only using normal human pharmaceutical preparations. For example many of the anabolic steroids used by athletes are approved for veterinary use only and since their metabolism is different in animals other than man, the human excretion processes have to be determined. The final stages in the detection process are where the analytical chemist's expertise comes to the fore. This involves selection of an extraction procedure, and instrumental process for the measurement of the desired analyte or analytes. Ideal procedures will incorporate high extraction efficiency for the compounds of interest as well as a selective and sensitive detection method.
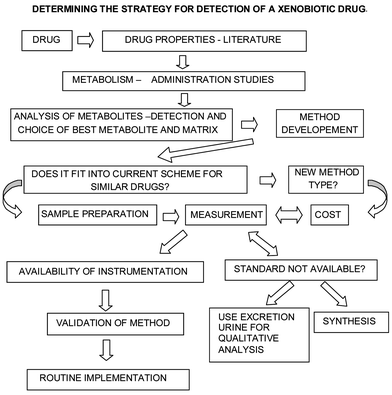 | ||
| Fig. 2 Schematic showing generalised process to be considered in determining a method of analysis for a doping substance. | ||
The laboratory product provided to the client requesting the drug test consists of two types of “tests”. These are “in-competition” (those collected immediately after competition events) and “out-of-competition” (those collected at any time at short or no notice). Analyses are performed to suit these categories. In-competition samples are analysed for the presence of all classes of banned drugs, whilst out-of-competition samples are analysed for steroids, diuretics, and HCG (in males only). The reason why in-competition and out-of-competition samples are treated differently relates to the way in which the different classes of drugs are used to provide a competitive advantage.
Laboratories have developed multi-analyte screens for classes of compounds. Table 1 shows the procedures used at the Sydney 2000 Olympic Games to detect the various classes of compounds. Most methods use gas chromatography–mass spectrometry for both screening and confirmation.
| Class of compound | Matrix | Chemical property exploited | Conjugation | Extraction | Derivative | Instrument used |
|---|---|---|---|---|---|---|
| Stimulants | Urine | Basic drug | None | Liquid–liquid extraction at high pHH | None | GC/NPD; GCMS full scan |
| Narcotics | Urine | Basic drug | Glucuronide/sulfate | Extractive alkylation or liquid–liquid extraction | Methyl | GCMS SIM |
| Diuretics/Masking agent | Urine | Acidic/basic | None | Extractive alkylation; SPE | Methyl; None | GCMS SIM; LCMSMS |
| Anabolic Agents | Urine | Neutral | Glucuronide | SPE or liquid–liquid extraction | Enol-TMS, TMS | GCMS SIM |
| Peptide hormones | Urine/blood | Large molecule | None | None/ultra-filtration | None | Immunoassay/isoelectric focusing–double blotting |
| Anti-oestrogenic compound | Urine | Neutral/acidic | Glucuronide | SPE | TMS | GCMS SIM |
2.1 Stimulants
Stimulants need only to be sought during in-competition testing as their effect is immediate and so a single dose can give a competitive advantage during an event.The development of the method currently used for the detection of stimulants illustrates how the analytical process was developed and refined. The examples of stimulants given in Fig. 3 show that several are amphetamine related and all are basic in nature and all contain nitrogen. Thus the extraction of such compounds from urine makes use of the basic nature of most of these drugs. A simple extraction into ether at pH>9.5 will provide an extract containing most of the stimulants which are volatile and not conjugated to glucuronic acid. Nitrogen containing “neutral” drugs will also be extracted. To detect the range of stimulants from the most volatile amphetamine to the least volatile strychnine requires a method that can separate and detect a very wide range of compounds. Early methods (pre 1970) used gas chromatography using a packed column and a flame ionisation detector. This is a non-selective detector whereby all compounds extracted from the urine would be detected making the nitrogen containing signals part of a forest of peaks. In 1972 Hewlett Packard released the thermionic specific detector (TSD or NPD). This detector, which contains a coated ceramic bead, gave a high signal response to only those compounds containing nitrogen or phosphorus. It was the development and application of this specific method by the late Professor Manfred Donike at Munich in 1972 that initiated modern doping control.6 With the later development of quartz capillary columns and the availability of bench-top mass spectrometers coupled to gas chromatographs (GCMS) for confirmation of suspicious results, the method is now easily capable of detecting and confirming the presence of stimulants in trace amounts in urine. Fig. 4 shows the NPD chromatograms of a typical blank athlete urine sample and a urine sample that contains excessive background, before and after spiking with 500 ng mL−1 of amphetamine and 200 ng mL−1 of strychnine. The typical blank urine sample is very clean only showing peaks for the internal standards and caffeine. The presence of the spiked drugs can clearly be seen at 1.1 and 5.8 minutes even in the “dirty” sample.
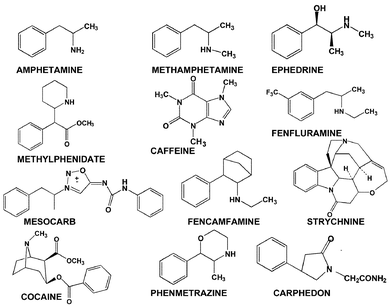 | ||
| Fig. 3 Structures of various stimulants banned in sport. | ||
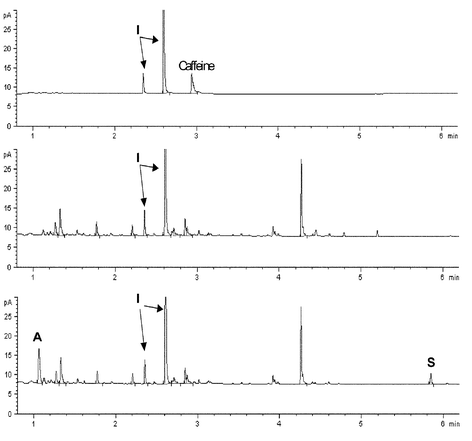 | ||
| Fig. 4 GC-NPD trace (6 minute run) of the extract from a typical blank urine (top), from a “dirty” urine (centre) and the same “dirty” urine spiked (bottom) with 500 ng mL−1 of amphetamine (A) and 200 ng mL−1 of strychnine (S). The internal standards are marked (I). | ||
2.2 Steroids
Anabolic steroids are included in both in and out-of-competition testing as they act, in conjunction with training, to increase muscle mass during a period well before an event or series of events. The increased muscle mass and resultant competitive advantage persist for several weeks after the anabolic steroid use has been discontinued and any trace of the compound administered may have disappeared. Thus if only predictable in-competition testing was used then the educated cheat will avoid detection. The most effective way to detect the use of drugs such as anabolic steroids, which have benefits that can extend beyond the period of analytical detectability, is with unannounced out-of-competition testing. To achieve the benefits of anabolic agents it is necessary to take a prolonged course during the training phase and the likelihood of being selected for testing at any time during steroid use is obviously disruptive to such a doping programme.The detection of anabolic steroids is a much more difficult process than the detection of stimulants. Whereas most stimulants are basic drugs containing nitrogen that allows detection using a selective NPD device, steroids do not have functionality that allows easy specific measurement. The anabolic steroids have structures related to testosterone and are neutral relatively non-polar compounds as shown in Fig. 5.
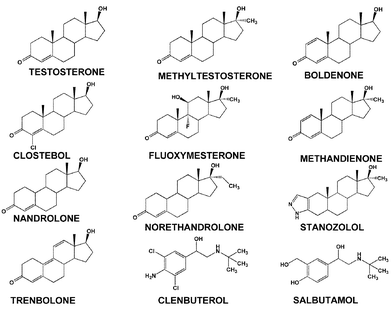 | ||
| Fig. 5 A selection of anabolic agents banned in sport. | ||
The problems relating to anabolic steroid detection are:
• The detection of very low levels (below 10 ng mL−1) is desirable for administered steroids because anabolic steroids assist the build up of muscle mass during the training period and hence can provide a performance advantage for some considerable time after their use has been stopped. Greater sensitivity equates to longer retrospectivity.
• High levels of naturally occurring steroids that require knowledge of population reference ranges and interpretation of an individual's acceptable range from longitudinal data.
• Steroids are frequently excreted as glucuronides or sulfates that must be hydrolysed prior to analysis.
• Metabolism of the anabolic steroids is complex and often only metabolites can be identified.
• Many of the steroids and particularly the hydroxylated metabolites are not amenable to analysis by GC without derivatisation to enhance their volatility.
The method used to detect anabolic steroids was pioneered by Professor Donike's group at the Cologne IOC laboratory. This required not only the development of hydrolysis and extraction procedures, but also the identification and synthesis of metabolites.7 This was facilitated by the development of and the synthesis of the efficient derivatising agent N-methyl-N-trimethylsilyltrifluoroacetamide (MSTFA) both used alone or with catalysts such as trimethylsilyl iodide (TMSI) or trimethylsilyl imidazole and which proved to be universally suitable for steroid analysis since it minimised formation of multiple derivatives.8
The use of mass spectrometry for drug detection was greatly improved by the availability of the bench-top quadrupole mass spectrometer coupled to capillary gas chromatography (GC/MS) in the early 80's. Routine large scale screening of samples for doping substances could be undertaken and was used very successfully for the first time during the 1984 Olympics in Los Angeles. This technique provided both the ability to identify metabolites in full scan mode and the ability to selectively detect them at low concentrations in selected ion monitoring (SIM) mode. During the 1980's the metabolism of many anabolic steroids was explored and detection capabilities improved.9 The classic case was the discovery of the hydroxylated metabolites of stanozolol10 thus allowing easy detection of its use. This capability was useful in providing the positive result against Ben Johnson during the Seoul Olympics in 1988.
The method used to extract and analyse anabolic steroids has changed little over the past 20 years since it was first demonstrated at the Cologne Workshops, and is shown in a schematic fashion in Fig. 6. It consists of a rapid cleanup on a solid phase material (e.g. XAD-1, C18), followed by enzyme hydrolysis of conjugated metabolites using a β-glucuronidase (e.g.E. coli or H. pomatia derived), extraction into an organic solvent and finally derivatisation using MSTFA/TMSI (stabilised with a thiol such as dithioerythritol or ethanethiol) and mass spectral analysis of the trimethylsilyl (TMS) and enol-TMS derivatives.11 Recent variations include direct hydrolysis of the urine sample followed by solid phase extraction (SPE) cleanup to replace solvent extraction. This later process is more amenable to automation using SPE workstations. It however suffers from some issues relating to direct hydrolysis on the urine where substances in the urine may interfere with the enzyme hydrolysis step or bacteria in the urine may metabolise some of the analytes during enzyme incubation step. These side processes can be readily monitored by using deuterated surrogate internal standards such as D4-androsterone glucuronide and D5-etiocholanolone to allow the hydrolysis and biological activities to be checked by simply looking at their ratios during data analysis and comparing to expected values. The use of isotopically labelled materials such as androsterone and etiocholanolone is just one example of the extensive quality assurance and quality control procedures that form an essential part of all the screening and confirmation procedures used for sports drug testing.12
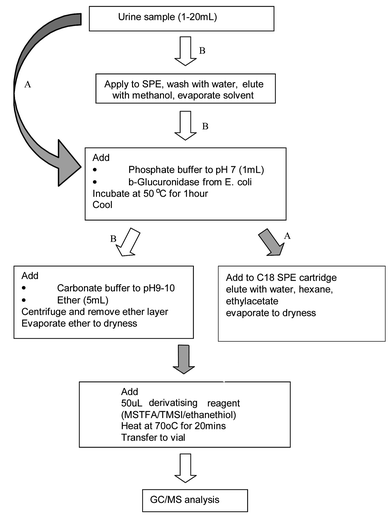 | ||
| Fig. 6 Sample preparation for anabolic agents. | ||
The GCMS instruments used for anabolic steroids analysis allow the steroids and their metabolites to have chromatographic and mass spectral data recorded. The data is most conveniently analysed by plotting individual characteristic ion chromatograms in narrow windows within the expected retention times. Use of the total ion chromatograms is difficult because of the very large background present that can envelop the peaks of interest for the compounds. By extracting plots of characteristic ions from the chromatographic data the background arising from all the other ions, which are also in the data set, is largely removed and the peaks may be more easily seen. By printing the data in a format that allows several ions for each substance to be lined up, visualisation of the presence or absence of anabolic agents or their metabolites is made routine. Software Macros for performing this process were written and distributed by the Cologne IOC accredited Laboratory in the early 80's using Hewlett Packard software. The basic techniques are still adopted by most laboratories regardless of the current instrumentation. Fig. 7 shows some typical data for spiked urine samples, demonstrating how the selected ion traces enable the detection of the compounds of interest.
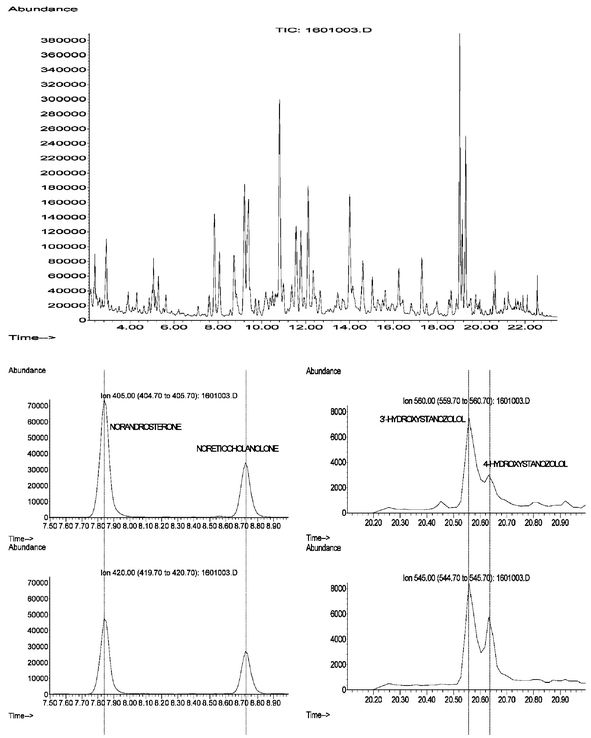 | ||
| Fig. 7 TIC (top) and ion chromatograms for 19-nortestosterone (nandrolone) metabolites, 19-norandrosterone and 19-noretiocholanolone (left) and for stanozolol metabolites 3′-hydroxystanozolol and 4β-hydroxystanozolol (right). | ||
In 1995 the Cologne laboratory published work indicating that lower detection limits and the retrospectivity of a number of anabolic steroids could be achieved by reanalysing the sample extracts using gas chromatography–high resolution mass spectrometry (GC/HRMS).13 Up until this time HRMS instruments had not been used for high volume routine analysis except in the measurement of dioxins and even then the samples presented to the instrument were very much cleaner than those obtained with derivatised urine extracts. The enhanced selectivity of the HRMS compared to the bench-top quadrupole instruments as well as increased sensitivity make this procedure quite effective. Using HRMS it became possible to routinely detect anabolic steroids at or below 2ng mL−1, whereas the bench-top GC/MS instruments, using the same extraction process, were limited to 10 to 20 ng mL−1 in most cases. While signal strength was sufficient in the quadrupole instrument the biological background often swamped the ions of interest. The HRMS instrument allows one to look below the noise by selecting the ions with greater precision. The quadrupole-based instrument has nominal unit mass resolution that means that for example a mass of 85 can be distinguished from a mass of 86. The HRMS can be set to resolutions ranging from 1000 to 15,000 when connected to a gas chromatograph. At a resolution of 6000 (10% valley) it is capable of distinguishing between masses of 86.0970 (C5H12N) and 86.1017 (C513CH13). The 86 ion containing nitrogen is the base peak of some beta-agonists, such as clenbuterol, when analysed as their TMS derivatives, whilst the carbon only 86 ion can come from any compound that contains a saturated hydrocarbon chain longer than six carbon atoms and is a very common source of background noise in real samples. At high resolution the background noise with mass other then 86.0970 ± 0.01 is greatly reduced and the selected ion chromatogram shows the peak of interest much more clearly. Fig. 8 shows the selected ion chromatograms for a real sample containing metabolites of stanozolol at a concentration of approximately 3ng mL−1. The normal quadrupole GC/MS data is equivocal at best because of the high background noise hiding the signal, whereas the HRMS data clearly shows the presence of the two characteristic stanozolol metabolite ions 545.3415 and 560.3650.
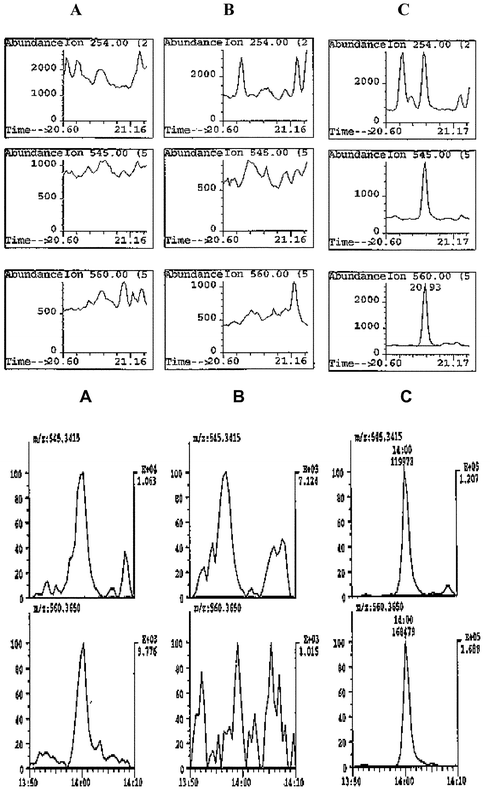 | ||
| Fig. 8 Selected ion chromatograms obtained on the same urine extract showing the ease of detection of the metabolite of stanozolol using HRMS (bottom) compared to quadrupole MS (top). A represents the sample, B the blank urine and C the pure metabolite standard. | ||
Improvements in ion detection have also been obtained by a number of laboratories using tandem mass spectrometry (GC-MS/MS). This technique enhances selectivity by isolation of an ion of interest (precursor ion) and then fragmenting it to produce the mass spectrum of the selected ion (product ions). Other substances of different chemical structure but with the same precursor ion mass will fragment differently and produce a different array of product ions, and hence will not be detected in the ion chromatograms produced for the drug being measured. In general this technique has a limit of detection close to the lower limit of the range required for steroid analyte measurement (1 to 2 ng mL−1). It is however particularly useful in confirming drugs present in a complex matrix by full scan measurement without the extensive purification otherwise needed. For example the TMS derivative of the main metabolite of stanozolol has a characteristic ion at 545 which when fragmented gives four characteristic ions at 455, 387, 277 and 439 all with abundances of at least 10%.14
One of the steps forward since 1990 in the analysis of anabolic steroids has been the availability of their metabolites as pure substances.7,15 Previously, the only source of such metabolites was from excretion studies performed by administering the drug of interest to a drug free volunteer and collecting urine samples at various time intervals before and after administration. These urine samples were analysed and the metabolites identified. These analysed samples became the reference materials for drug detection. From large synthetic programmes standards are now available which can be analysed at the same time as the blank urine and the sample and comparisons made.
Other anabolic agents such as the β2-agonists clenbuterol and salbutamol are also banned. These substances have been shown to enhance the development of lean muscle in animal studies and are thereby banned in all meat products by the European Union. This work together with the findings that athletes were using clenbuterol for its anabolic enhancement properties have prompted the banning of their use. They are readily detected by GCMS analysis either in the routine steroid procedures or as part of the beta-blocker analysis (see 2.4) as TMS, trifluoroacetyl (TFA) or TMS/TFA derivatives. Clenbuterol has a very low required detection level due to the low dose used but can be readily measured using HRMS or MSMS screening techniques at 2 ng mL−1 or less in routine work.
Salbutamol is more problematic in that use is permitted by inhaler to treat asthma but oral administration is banned. The laboratory is required to distinguish between oral and inhaled routes of administration. This has been attempted by measuring the difference between the R and S enantiomers16 because there is a difference in metabolism of the optical forms depending on the routes of administration. Further work has also shown that if salbutamol is given in reasonable therapeutic doses by inhalation, the urinary level remains below 500 ng mL−1.17 This research has led to a threshold of 1000 ng mL−1 being set above which it is deemed that the consumption is not by a permitted route and is likely to have been oral administration and for the purpose of doping. Athletes wishing to use salbutamol need to get prior permission from a medical panel within the sporting federation. Urinary levels below the threshold are considered to arise from use by non asthmatics for stimulant effects unless documented evidence of medical need is provided. The usual scenario is that when salbutamol is found in the urine at a level below 1000 ng mL−1 and is reported, a check is then made on the medical status and permissions. If the athlete has permission or can show an asthma-related condition requiring the use of a beta-agonist, no further action is taken.
Anti-estrogenic substances have recently been added to the IOC/WADA list. These are taken to counteract the effects of anabolic steroid abuse by: a) stopping the minor metabolic pathway of the anabolic steroids leading to estrogens which give rise to side effects such as breast development in males; b) stimulating the hypothalamus to release hormones which stimulate the testes to produce testosterone – a major effect of administered steroids on the testes is for them to rapidly cease to function normally towards androgen biosynthesis and to reduce markedly in size. The anti-estrogenic drugs include clomiphene, tamoxiphen and cyclofenil as examples of aromatase inhibitors that are banned in males. These are detected in the same screen as the anabolic agents.
2.3 Diuretics
Diuretics are banned for two main reasons. The first is their ability to assist rapid water loss as urine and thus attaining a reduction in weight. This is useful in weight class sports where the drug may be taken a little time before the weighing process, which is done prior to competition. The athlete eliminates water and at weighing attains a lower weight class (a weight they may have trouble attaining by natural means). After the weighing re-hydration leading up to the event brings them back to their higher natural weight but allows them to compete against lighter opponents. The second reason results from the ability of diuretics to rapidly dilute urine by increasing renal flow. This can make it more difficult for the laboratory to detect banned drugs especially at lower levels towards the end of the drug excretion process. The carbonic anhydrase inhibiting diuretics also will make the urine alkaline thus reducing the excretion of basic drugs.Diuretics are frequently included in the out-of-competition drug screen because of their ability to dilute the urine and make the detection of drugs more difficult. They are mostly polar substances that contain aminosulfonyl substituents or carboxylic acid moieties as shown in Fig. 9. These make them relatively water soluble and involatile. Many laboratories therefore used high performance liquid chromatography (HPLC) with diode array detection (DAD) as a method of choice. This procedure suffers from the wide range of polarities of these substances and for many diuretics the urine background is difficult to eliminate sufficiently to give detection levels that may be required for a multi-residue analysis. The DAD detection technique is not specific enough to allow unequivocal identification of the substances and a mass spectral methodology is required for confirmation.
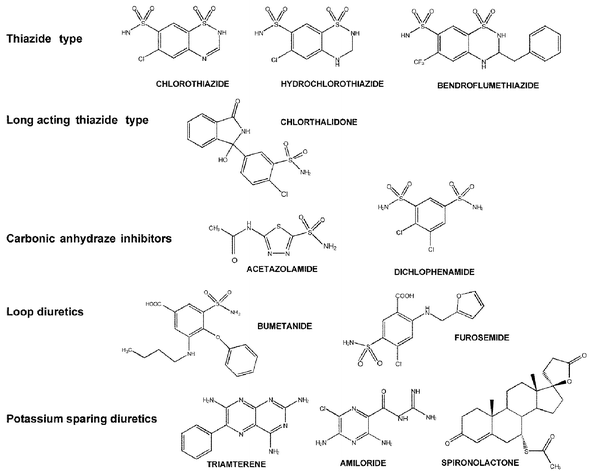 | ||
| Fig. 9 Examples of types of diuretics showing the variety of functionality in the molecules. | ||
The confirmation technique most widely used required methylation of the aminosulfonyl group followed by GCMS measurement. This methylation could be achieved by two main processes – direct methylation using methyl iodide/potassium carbonate in acetone at 60°C or by extractive alkylation where the diuretics are extracted and derivatised in a one step process using a phase transfer reagent with methyl iodide in an organic solvent such as toluene.18 The methylated products are reasonably volatile and can be analysed by GC/MS. The extractive alkylation process is shown in Fig. 10 and it can be used to both screen for diuretics, giving lower detection limits and better selectivity than the HPLC DAD method, as well as for confirming them.
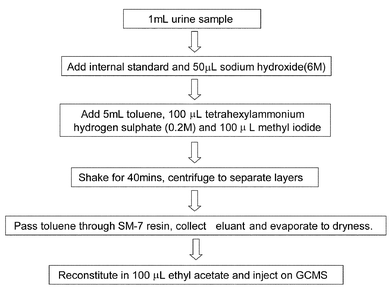 | ||
| Fig. 10 Scheme for preparation of sample analysis of diuretics by extractive alkylation. | ||
The advent of relatively inexpensive and robust electrospray mass spectrometric detectors for HPLC has seen a strong movement towards a relatively simple analysis of polar substances without the need for derivatisation. The technique for diuretics is best used in the MSMS mode using both positive and negative ions which gives high selectivity and sensitivity and is becoming the technique of choice for analysis of diuretics. It is amenable to high throughput multi-residue screening for a very large array of diuretics.19
2.4 Narcotics
These substances appear to have been abused in sport prior to 1980. Their usefulness in sport is limited but may be an issue where pain relief has a serious bearing on performance. Their side effects of constipation and drowsiness do not help in most sports. They are however an important issue within world health where their use is controlled by international agreements and thus they remain part of the banned drugs suit.The detection of narcotics has not been difficult as they are relatively easily extracted and analysed by GC using either a specific detector such as the NPD or by GCMS. Many narcotic substances are excreted as free drug together with glucuronide or sulfate conjugates. The general extraction process usually allows them to be analysed together with hydroxylated stimulant metabolites (e.g. pholedrine, hydroxyamphetamines) and can use either enzyme or acid hydrolysis of the conjugates followed by extraction of the drugs using an organic solvent at pH 9.5. The final extracts can be derivatised in a number of ways – trifluoracetates for GC/NPD or GCMS screening, TMS derivatives for GCMS screening and TMS/TFA derivatives for GCMS screening.20 The latter is particularly useful for substances with hydroxy groups as well as primary or secondary amino functions.
In some laboratories the steroid procedure is used for narcotics screening. The diuretics extractive alkylation method using tert-butyl methyl ether as solvent is also highly effective for a wide range of phenolic stimulants, their metabolites and a good range of narcotic substances.
2.5 Beta-blockers
Beta-blockers are banned because of their ability to slow the heart rate and thus provide the athlete with the ability to reduce tremor. This makes beta-blockers particularly useful in sports such as shooting, archery and snooker. In some multi-disciplinary sports such as modern pentathlon judicious ordering of the events in order to have an active event immediately after the shooting makes the use of beta-blockers less attractive.The analysis of beta-blockers is similar to that used for narcotics and steroids. Enzyme hydrolysis of the urine using Helix pomatia to deconjugate sulfates and glucuronides or Escherichia coli for cleavage of glucuronides, give the free drug or metabolite which can then be purified by solvent extraction or SPE cleanup. The residue from the cleanup is derivatised to provide either a TMS derivative in a similar fashion to the steroids method or by using MSTFA and N-methyl-bis-trifluoroacetamide (MBTFA) to provide the O-TMS/N-TFA derivatives. A multi-residue analysis using GCMS can then be used to detect the individual substances. Recent work on using LCMS has provided a good means of detecting these substances.21
2.6 Endogenous and exogenous compounds
The screens discussed so far have mainly involved the detection of pharmaceutical compounds that do not occur naturally in the body. With such exogenous compounds, detection and proof of identity at any level can lead to a doping violation. There are a few exceptions made for commonly used drugs such as pseudoephedrine where the level above which a doping violation has occurred is set at >25 μg mL−1, considerably above the detection level. However for most exogenous compounds, the full power of the analytical chemist's repertoire can be brought to bear and in general the detection of such compounds is relatively straightforward. Compounds that occur naturally in the body present a markedly different problem.Mere detection of an endogenous compound is obviously no proof of doping. It might be thought that detection of an elevated level of an endogenous compound could be useful in doping control but determination of what is an “elevated” level is not straightforward. Many physiological parameters such as the haemoglobin concentration in the blood have a relatively narrow range within a given individual and hence a relatively small change may be perceived as abnormal. Samples that arrive at a sports drug-testing laboratory are identified solely by a number and could theoretically come from an athlete of any ethnic background from any country. Thus the only way of establishing deviation from normality is to use the range for the entire world population which is very much larger than for a single individual. In general this means that it has not been possible to establish levels which can effectively define doping with endogenous compounds.
When testosterone is injected the T/E ratio increases not only because testosterone is excreted due to the material injected but also because the natural production of both testosterone and epitestosterone is suppressed. Should a T/E ratio greater than 6 be detected, an investigation has to be carried out that includes reviewing previous test results and/or carrying out additional testing over the next three months or so. The purpose of this testing is to determine if the T/E ratio is varying, which indicates administration, or is relatively constant as in the case of a naturally elevated T/E ratio due to suppressed epitestosterone excretion. Despite levels above 6 being considered an offence a few subjects have been found with natural levels exceeding this threshold, usually due to low epitestosterone excretion. Monitoring the T/E values for individuals shows that the values do not vary over time by more than ± 30%. Fig. 11 shows schematically the variation of T/E over time and includes a normal subject with a T/E close to 1, a subject with a naturally elevated T/E, and someone who has been doping with testosterone and stops after being advised that they are under investigation. This is one case where stopping taking the drug makes it easier to confirm its use. It could be argued that a smart athlete would continue taking testosterone and maintain an apparently natural elevated T/E. This has been attempted but it is difficult to achieve the required constancy of T/E ratio. It is just such a possibility of continued low dose administration, along with the desire to obtain a definitive result in less than three months, that has driven researchers to consider the use of another advanced analytical technique namely gas chromatography carbon isotope ratio mass spectrometry (GC-CIRMS).
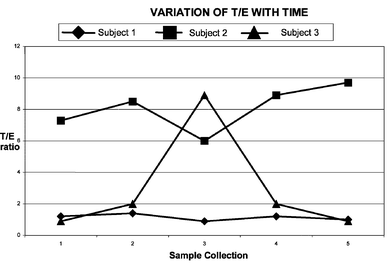 | ||
| Fig. 11 Schematic representation of longitudinal variation for the T/E ratio in cases of individual with normal values, naturally elevated, and having administered testosterone before the third collection time point. | ||
CIRMS has been extensively used for detection of adulteration in food and in geochemical analysis.23 In this technique the compounds of interest are separated by gas chromatography and then combusted to carbon dioxide in a furnace. The carbon dioxide is passed into a small magnetic sector mass spectrometer that simultaneously measures the ions of mass 44 (12C16O16O), 45 (13C16O16O), and 46 (13C16O18O) using fixed Faraday cups. By precisely measuring the ratio of mass 45 to 44 it is possible to measure the amount of carbon 13 relative to carbon 12 in the compound being analysed. Carbon isotope ratios are expressed in terms of δ13C values, with units in per mille (‰), where:
| δ13C = [(rsample/rstd) − 1] × 1000 |
Measurements are expressed on a scale where the zero point is set by the isotopic composition of Pee Dee Belemnite, a marine carbonate that is relatively enriched in 13C. This results in most δ13C values being negative. Results are expressed in per mille because the measurements are generally very small; a difference of 1‰ on the δ13C‰ scale corresponds to a change in 0.001099 atom% in natural 13C abundance.24 Measurements made with CIRMS have a precision better than 0.5‰.
All living matter is derived through photosynthesis from CO2 in the atmosphere, where δ13C = −7‰, or in marine environments where δ13C = 0‰. Plants with C-3 and C-4 metabolism pathways discriminate to different degrees against 13CO2 and the isotope ratios in biochemicals from plants reflect the biochemical grouping of the plants.25 The carbon isotopic composition in humans reflects the diet. This is why human breath samples of Americans, who eat a lot of corn and corn products (C-4 plants), are enriched in 13C compared with Europeans, whose diet is more wheat based (C-3 plants). Isotopic composition has been found to be useful in a sports drug-testing context because synthetic steroids are usually derived from stigmasterol obtained from soy, a C-3 plant which gives synthetic testosterone δ13C values in the range −25.9‰ to −32.8‰.26 Synthetic copies of other endogenous steroids such as androstenedione are similarly depleted in 13C. Steroids produced naturally within the body have delta values that reflect the average carbon isotope ratio of the food consumed by the subject. The difference in carbon isotope ratio between the natural and synthetic steroids, though small, is statistically significant, and measurable.
In urine it is possible to find steroidal compounds which are unaffected by synthetic steroid administration and these “background markers” can be used to determine whether administration of synthetic steroids has taken place. Administration of synthetic endogenous steroids leads to a measurable depletion of 13C content in the parent compound, and also in its metabolites, whilst the background markers are unaffected.27 Cases where the δ13C values of steroid metabolites are greater than four units more negative than those of the marker compounds are highly indicative of doping. The introduction of CIRMS into sports doping detection means not only that there is now an independent confirmation for elevated T/E results but also that it is now possible to confirm doping with synthetic endogenous steroids such as testosterone without having to wait a considerable period for follow up testing to be completed. CIRMS was used for the first time at a summer Olympic Games in Sydney 2000. Whilst no cases were detected during this event, three cases of testosterone doping were detected by elevated T/E ratios in the subsequent Paralympic Games. For the first time at a major event it was possible to confirm these results using CIRMS and immediate penalties were imposed.28
Considerable effort has been put into the detection of these drugs and this justifiably reflects the difficulty of the problem. Most banned peptide hormones are naturally occurring substances some with varying diurnal variation in blood levels. Many are not efficiently eliminated in the urine so there has been the need for the implementation of blood collection as well as urine. Professor P. Sonksen undertook the problem of detection of the use of growth hormone (GH) as part of an EU and IOC funded grant. This work concentrated more on the identification of markers of use rather than the administered substance. The investigation focused on the change induced in the endogenous substances that are part of the activity mechanism of the body. Effects on insulin like growth factor (IGF-1) and its binding proteins, and on marker proteins for bone and connective tissue turnover were measured before and after administration and compared for statistical significance.29
A similar approach was undertaken by our group together with the Australian Institute of Sport and other international collaborators for the detection of EPO in blood (EPO2000 project). Parameters such as haemoglobin, haematocrit and % reticulocytes were measured using a commercial haematology analyser supplied by Bayer. The immunoassays for soluble transferin receptor (sTfr) and EPO in the serum were also performed using proprietary analysers. These data were used to construct multivariate models which gave a good indication of recombinant EPO (rhEPO) use.30 The Paris IOC accredited laboratory took a different approach. They were able to show separation of EPO isoforms occurring in urine samples by an ingenious process of iso-electric focusing followed by a double blotting procedure.31 The second blot is an image of the EPO isoforms and is visualised by chemiluminescence. There was a marked difference between normal EPO produced in the body and recombinant EPO in products such as Eprex™. Recent development of a modified EPO type drug – Aranesp™ (darbepoietin alfa) – has produced a substance with a very long half-life compared to EPO. This drug affects blood parameters in a similar way to EPO and is easily detectable as a series of bands more acidic than urinary EPO in the double blotting technique.
Considerable anti-doping research is being directed towards the detection of peptide hormones. There are difficult issues to be overcome in the detection of these compounds and this poses both technical and philosophical problems. Most of the technical problems can be overcome albeit at great cost and effort but unless the philosophical problems are addressed then drug testing for some hormones will remain ineffective even when analytical methods are available. The major philosophical problem arises from the desire of those enforcing infringements of anti-doping codes to accept only direct tests as proof of doping. For example in August 2000 at a special meeting convened by the IOC to consider the results of the EPO2000 project a proposal was made to accept the results of one or more statistical models based on whole blood and serum parameters as proof of doping with EPO. The cutoffs proposed were based on thousands of samples collected from some 1200 elite athletes in 12 countries around the world.32 The blood parameters were well able to detect the use of rhEPO for most of the period of use and also well after administration had ceased. Results from the direct urine test were also presented. The expert group concluded that the indirect blood test could not be used for sanction but should be used to support the urine findings and indicate athletes who should be subjected to further unannounced testing by the direct urine test. However this approach has a major limitation in that it assumes that a useful direct test for the drug or its metabolites will always be available. Fortunately, at present the direct test using urinary detection of EPO is suitable for catching cheats in some situations.
Despite the protein sequence in EPO being cloned from the human gene and hence being identical to that found naturally, the extensive glycosylation of the protein is slightly different in the recombinant product, which is produced in Chinese hamster ovary cells, than in the material produced naturally by the human kidney. This difference in glycosylation is the basis of the direct urine test for EPO doping. Fig. 12 shows the difference in isoform patterns for recombinant and urinary EPO. However this test will cease to be effective if a recombinant EPO is made with glycosylation more similar to that found naturally. Even if such a recombinant EPO is not made there is a very real likelihood that means will be found to stimulate the kidney or other organs by gene implantation to produce more EPO.33 Once either of these objectives has been achieved then there will be no longer a direct test for EPO and hence no test for EPO, unless indirect testing is accepted. Currently the best situation is to collect both blood and urine and to measure all parameters for both matrices. From the urine test it will be possible to report recent rhEPO use but otherwise the blood parameters will provide considerable intelligence information about those who may be or are abusing the drug so they can subsequently be targeted for unannounced testing. The special problems associated with effectively detecting and deterring EPO doping using both blood and urine testing have recently been considered.34 Only by using such a holistic approach can the cheats be detected.
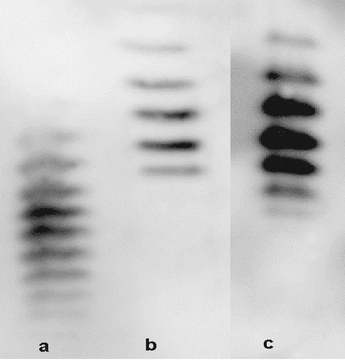 | ||
| Fig. 12 Western blots of (a) normal urinary EPO, (b) recombinant EPO, and (c) EPO recovered from the urine of a subject after recombinant EPO injections (cathode top of gel and anode bottom of the gel). | ||
As yet there is no accepted doping test for growth hormone (GH) either direct or indirect. The problems are even greater with developing a direct test for GH. Since circulating GH includes several isoforms with approximate molecular weights of 22 kDa, 20 kDa and 17 kDa.35 any direct test for growth hormone will probably be based on the measurement of the growth hormone isoform ratios. The recombinant product is exclusively 22 kDa and hence those injecting recombinant GH will have predominantly 22 kDa GH circulating in their system. However whilst some hormones such as EPO have relatively small diurnal variations the release of GH is pulsatile with blood levels varying by three or four orders of magnitude several times a day depending on food intake and physical activity. Thus it would be feasible for an athlete injecting GH to have a high 22 kDa to 20 kDa ratio shortly after injection but this ratio would be expected to reduce after heavy exercise. Thus a difference of only a few hours in the timing of the blood collection could significantly affect the result. To overcome this variability many GH clinical studies have used pooled multiple blood collections. However this will not be possible in the normal sports drug-testing context.
Even if direct tests can be made to work there will always be the problem with most peptide hormones that the beneficial effect lasts far longer than any trace of the drug. Thus testing at competitions will only catch the naive drug user and cannot give a true picture of the extent of doping. Indirect tests for EPO and GH are likely to have much longer detection windows and hence be generally more effective both in-competition and out-of-competition. However these indirect tests are based on the statistical analysis of groups of users and non-users and there is a measurable, albeit low, probability of there being a false positive. If the medical history of individual athletes were to be recorded and used to monitor marked physiological changes, then indirect testing becomes simpler and more reliable, since the use of individual statistics is much better than population statistics for drug monitoring. The World Anti-Doping Agency has stated that it plans to introduce such a program in the future.
The development of direct tests for peptide hormones presents special problems for the drug-testing laboratory. Up until relatively recently most of the banned substances have been of relatively low molecular weight and amenable to analysis by gas chromatography with confirmation by mass spectrometry. The IOC code requires that confirmation of identity be by mass spectrometry although peptide hormones have been specifically excluded from this requirement. However the recent advances in biological mass spectrometry using atmospheric pressure ionisation (API) and matrix assisted laser desorption ionisation (MALDI) mass spectrometry, for which the 2002 Nobel prize in Chemistry was awarded, has made the mass spectral identification and characterisation of peptide hormones possible. The goal is now to apply these techniques to detect and confirm the presence of banned hormones in relatively small blood or urine samples where the total amount of material available may only be a few femtomoles. Thus sports drug testing laboratories now face the prospect of developing and applying completely new methodologies which require not only very large capital expenditure and high ongoing costs, but also the development of new skill and expertise.
2.7 Enhancement of oxygen transfer
There are number of methods which can be used to artificially increase an athlete's normal oxygen carrying capacity. These range from small molecules such as RSR13 that are readily detectable in urine36 to reinfusing the athlete's own stored blood (often referred to as blood doping) a process that is essentially undetectable. The most popular means at present of enhancing aerobic performance is the use of recombinant EPO that has been discussed above. However as the testing for EPO improves it is likely that those athletes who cheat will look elsewhere for a performance benefit.Blood substitutes are a likely source of such benefit. These products are under development by several companies as an alternative to blood transfusions because of the problem of obtaining sufficient blood to meet demand coupled with the difficulty in ensuring that the blood is not contaminated or infected by an increasing variety of lethal viruses. They are designed to be used in surgical operations and emergencies in place of donated blood.37 The products which are most advanced in their clinical trials are based on cross-linked and polymerised haemoglobin. They do not, and are not intended to, carry out all the functions of whole blood but make up volume after blood loss and deliver oxygen to the tissues. In fact it has been shown that a haemoglobin based blood substitute or haemoglobin based oxygen carrier (HBOC) has a higher oxygen carrying capacity per unit volume than whole blood.38 This means that an athlete who infused an HBOC would significantly increase their aerobic performance. The beneficial effect of such an infusion would be virtually instantaneous whereas to achieve an effect with EPO would take at a minimum two weeks.
The HBOCs that are in active development are all based on haemoglobin, either bovine (Hemopure™) or human (Hemolink™ and PolyHeme™), that has been chemically modified to overcome the toxic effects that accompany the presence of high concentrations of free haemoglobin in plasma.39 The modifications are intended to markedly increase the molecular weight and size of the molecule and typically involve crosslinking of the alpha and beta subunits of haemoglobin coupled with polymerisation or conjugation to another large molecule. In normal blood, haemoglobin is contained within the red cells and there is very little extracellular haemoglobin. The presence of large amounts of highly coloured “haemoglobin” in plasma could be indicative of the use of an HBOC. Haemolysis of the blood either during or after collection could lead to significant quantities of haemoglobin being present in the plasma. In our laboratory's experience such haemolysis is relatively infrequent and so a simple visual inspection of the colour of the plasma is a good first screen for detecting the abuse of HBOCs. Of course some means of distinguishing natural haemoglobin arising by haemolysis from the chemically modified HBOC is required. One way of achieving this is by the use of size exclusion chromatography because HBOCs have a much higher molecular weight than haemoglobin. An example of this is shown in Fig. 13. The characterisation of the HBOCs is also possible using electrospray mass spectrometry of enzymic digests to detect the differences between natural and cross-linked haemoglobin.40
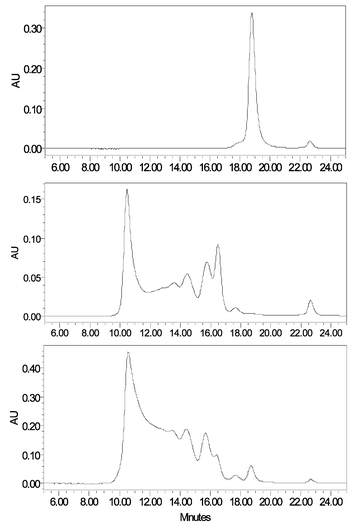 | ||
| Fig. 13 Size exclusion chromatography of human haemoglobin (top trace), 6.5 g L−1 of Hemopure™ in blank plasma (middle trace) and plasma from a human subject given Hemopure™ using a Bio-Sil SEC-250 Gel Filtration 300 m × 7.8 mm id column and a Waters Alliance 2690 HPLC system at a wavelength of 424 nm run with a magnesium chloride/bistris/EDTA buffer.40 | ||
At present it is normal to collect only urine samples with occasional blood samples for rhEPO testing. Unless blood samples are routinely collected to enable screening for several peptide hormones, HBOCs will not be detected. Whilst the planning and timing of the collection of the samples is not within the province of the laboratory, the laboratory should work with the collecting authority to provide valuable information as to when samples might be most effectively collected. For drugs which have short lasting effects comparable to their lifetime in the body the timing of sample collection is not critical and in-competition testing is required. The HBOCs (together with narcotics, diuretics, beta-blockers and stimulants) fall into this category. For the drug classes such as the anabolic steroids and EPO, the beneficial effect such as increased muscle mass or red blood cells, persist long after any trace of the drug used to gain the advantage has been eliminated by metabolism and excretion. Here testing at competition has a deterrent effect but is of little value catching the knowledgeable user.
Only one HBOC (Hemopure™) has so far been approved for human use, and then in very limited circumstances, so it might be felt that athletes would not yet have access to such materials. However for the last two years an HBOC (Oxyglobin™) has been approved for use in dogs in the USA and since it is common for athletes to use veterinary preparations it may have been tried as a doping agent.
3 Conclusions
The detection of performance enhancing drugs in sport is a specialised task within analytical chemistry that stretches the practitioner both by the scope of analytes that need to be considered and by the low detection limits that are frequently required. Until recently the primary expertise resided in the extraction and detection of relatively low molecular weight drugs using techniques such as gas chromatography mass spectrometry. With the advent of biopharmaceuticals there is a need to expand to include the detection and confirmation of peptide hormones and other macromolecules that are closely related or identical to compounds occurring naturally in the body. For some of these peptides such as EPO, methodology grew from research undertaken only after the use of rhEPO had become endemic in some areas of sport, notably professional cycling. The advent of the World Anti-Doping Agency has seen the creation of a well-funded research effort that should facilitate the development of methods before new drugs are readily available. This will always be particularly difficult when the compound being tested occurs naturally in the body. The banning of a compound or method does not mean that detection is currently possible. Research on the detection of growth hormone has been carried out over several years but as yet there is no validated test that can be used to detect its use for doping. Research on the detection of gene doping is only now being considered and unless the compounds produced by the implanted cells are somehow different from the endogenous material then detection other than possibly by biopsy will be extremely difficult.Good cooperation at an early stage between the laboratories, which need to develop tests, and the manufacturers is necessary to prepare for drug detection before athletes are found to be using the products for doping. Some pharmaceutical manufacturers, for commercial reasons, are reluctant to provide standards of their drugs while still in clinical trials. Unfortunately athletes have been found with pharmaceutical substances before they have been officially released. The advent of the WADA should facilitate this information transfer.
The need for extensive cooperation with all types of sporting authorities has been evident during the last year with the identification by the UCLA laboratory of athletes’ use of designer drugs using the 18-methyl series of anabolic agents such as gestrinone (Fig. 14). These include norbolethone41 and more recently its reduced analogue tetrahydrogestrinone (see http://www.usantidoping.org/files/pressRelease_10_16_2003_275.pdf), the data for which has yet to be published by the UCLA laboratory headed by Prof. D. Catlin. The discovery of athletes’ use of these substances, some of which have never been trialed in any in vivo test, animal or human, has highlighted the fact that conspiracies are occurring albeit not commonly, and that the ethical and safety values of some persons associated with sport are highly questionable.
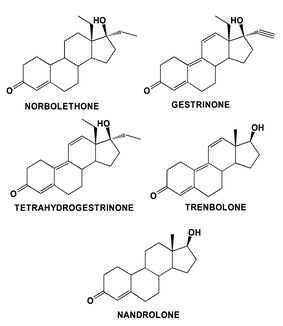 | ||
| Fig. 14 The structures of designer drugs showing the similarity to known anabolic steroids such as gestrinone, trenbolone and nandrolone. | ||
Many new techniques used lead to a further serious problem for drug testing authorities, namely cost. When a new stimulant or anabolic steroid was discovered it was a relatively inexpensive exercise to study its metabolism and include the compound in an existing analytical screen. However it is now necessary to develop a completely new analytical method for each different peptide hormone. The escalation in cost can be exemplified by the urinary EPO test that alone costs significantly more to perform than all the other current drug screens combined. To test all urine samples for the presence of recombinant EPO would more than triple the cost of sports drug testing worldwide. The approach adopted by the Australian Sports Doping Agency (ASDA) has been to target selected endurance sports (those most likely to benefit from EPO) for unannounced out-of-competition testing at those times when EPO would most likely be used. This approach has kept the cost down to a manageable level but has still needed special funding.
Any future test for growth hormone will also use a completely new methodology, further adding significantly to the cost of drug testing. As individual laboratory test costs increase then so does the overall cost of drug testing. Laboratories and sports administrators have limited budgets and must decide how to allocate their funds to best achieve their objectives. Unless there is a massive increase in funding it will not be possible to test all samples for all drugs, an approach which has been the required practice up until now. Not all current IOC/WADA accredited laboratories will be in a position to provide comprehensive testing and specialist WADA accredited laboratories will emerge. In time the massive expenditure that is currently being made in the field of proteomics, where more and more effort is being put into the rapid analysis of peptides and proteins, may help alleviate the funding situation. This may lead to the availability of reasonably priced and versatile equipment that can be applied to the detection of large biomolecules in sports drug testing.
Acknowledgements
We wish to thank the staff of ASDTL and IOC accredited laboratories for continuous development of methods reported in this review. We also wish to thank both the WADA and the Sport and Recreation (anti-doping) Program of the Department of Communications, Information Technology and the Arts for provision of research funding. We acknowledge the generous provision of HBOC samples (Hemopure™ and Oxyglobin™) and standards from the Biopure Corporation.References
- Minutes of the 67th Session of the International Olympic Committee, Mexico (IOC Lausanne), 1968, A11, p. 12 Search PubMed.
- L. Bowers, Clin. Chem. (Washington, D. C.)., 1997, 43, 1299 CAS.
- D. H. Catlin, C. K. Hatton and S. H. Starcevic, Clin. Chem.(Washington, D. C.)., 1997, 43, 1280 CAS.
- Code: http://multimedia.olympic.org/pdf/en_report_21.pdf and List: http://multimedia.olympic.org/pdf/en_report_542.pdf (cited 24/06/2003); from 2004 see the WADA website at http://wada-ama.org.
- ISO/PAS 18873:1999, International Organization for Standardization.
- Obituary for Manfred Donike in: Recent Advances in Doping Analysis (4) , W. Schänzer, H. Geyer, A. Gotzmann and U. Mareck-Engelke (eds.), Sport und Buch Strauss, Cologne, 1997, 8 Search PubMed.
- W. Schänzer and M. Donike, Anal. Chim. Acta, 1993, 275, 23 CrossRef.
- M. Donike and J. Zimmermann, J. Chromatogr., 1980, 202, 483 CrossRef CAS.
- R. Masse, H. Bi, C. Ayotte, P. Du, H. Gelinas and R. Dugal, J. Chromatogr., 1991, 562, 340.
- R. Masse and C. Ayotte, J. Chromatogr. Biomed. Appl., 1989, 497, 17 CrossRef.
- M. Saugy, C. Cardis, N. Robinson and C. Schweizer, Baillieres Best Pract. Res. Clin. Endocrinol. Metab., 2000, 14, 111 Search PubMed.
- C. Jimenaz, R. Ventura, X. De la Torre and J. Segura, Anal. Chim. Acta, 2002, 460, 289 CrossRef CAS.
- W. Schänzer, P. Delahaut, H. Geyer, M. Machnik and S. Horning, J. Chromatogr., B: Biomed. Appl., 1996, 687, 93 CrossRef CAS.
- J. Munoz-Guerra, D. Carreras, C. Soriano, C. Rodriguez and A. F. Rodriguez, J Chromatogr., B: Biomed. Appl., 1997, 704, 129 CrossRef CAS.
- S. Westwood, D. Hancock, C. Moule, B. Noble and S. Starling, Recent Advances in Doping Analysis (8), W. Schänzer, H. Geyer, A. Gotzmann and U. Mareck-Engelke (eds.), Sport und Buch Strauss, Cologne, 2000, 109 or NARL website http://www.agal.gov.au/NARL/refmaterials.html (accessed 20/06/2003) Search PubMed.
- R. Berges, J. Segura, X. De la Torre and R. Ventura, J. Chromatogr., B: Biomed. Appl., 1999, 723, 173 CrossRef CAS.
- R. Ventura, J. Segura, R. Berges, K. D. Fitch, A. R. Morton, S. Berruezo and C. Jimenez, Ther. Drug Monit., 2000, 22, 277 CrossRef CAS.
- A. M. Lisi, R. Kazlauskas and G. J. Trout, J. Chromatogr., B: Biomed. Appl., 1992, 581, 57 CrossRef CAS.
- D. Thieme, J. Grosse, R. Lang, R. K. Mueller and A. Wahl, J. Chromatogr., B:Biomed. Appl, 2001, 757, 49 CrossRef CAS.
- D.-S. Lho, J.-K. Hong, H.-K. Paek, J.-A. Lee and J. Park, J. Anal. Toxicol., 1990, 14, 77 CAS.
- M. Thevis, G. Opfermann and W. Schänzer, Biomed. Chromatogr., 2001, 15, 393 CrossRef CAS.
- J. Baenziger and L. Bowers, Recent Advances in Doping Analysis,M. Doniker, H. Geyer, A. Gotzmann, U. Mareck-Engelke and S. Rauth (eds.), Sport und Buch Strauss, Cologne, 1994, p. 41 Search PubMed.
- W. Meier-Augenstein, LC-GC, 1997, 17 Search PubMed.
- W. Brand, J. Mass Spectrom., 1996, 31, 225 CrossRef CAS.
- B. N. Smith and S. Epstein, Plant Physiol., 1971, 47, 380 CAS.
- M. Ueki and M. Okano, Rapid Commun. Mass Spectrom., 1999, 13, 2237 CrossRef CAS.
- C. H. L. Shackleton, A. Phillips, T. Chang and Y. Li, Steroids, 1997, 62, 379 CrossRef CAS.
- R. Kazlauskas, Clin. Biochem. Rev., 2002, 23(ii), 35 Search PubMed.
- J. D. Wallace, R. C. Cuneo, R. Baxter, H. Orskov, N. Keay, C. Pentecost, R. Dall, T. Rosen, J. O. Jorgensen, A. Cittadini, S. Longobardi, L. Sacca, J. S. Christiansen, B.-A. Bengrsson and P. H. Sonksen, J. Clin. Endocrinol. Metab., 1999, 84, 3591 CAS.
- R. Parisotto, M. Wu, M. J. Ashenden, K. R. Emslie, C. J. Gore, C. Howe, R. Kazlauskas, K. Sharpe, G. J. Trout, M. Xie and A. G. Hahn, Haematologica, 2001, 86, 128 Search PubMed.
- F. Lasne, L. Martin, N. Crepin and J. de Ceaurriz, Anal. Biochem., 2002, 311, 119 CrossRef CAS.
- K. Sharpe, W. Hopkins, K. R. Emslie, C. Howe, G. J. Trout, R. Kazlauskas, M. J. Ashenden, C. J. Gore, R. Parisotto and A. G. Hahn, Haematologica, 2002, 87, 1248 Search PubMed.
- A. Bartholomew, S. Patil, A. Mackay, M. Nelson, D. Buyaner, W. Hardy, J. Mosca, C. Sturgeon, M. Siatskas, N. Mahmud, K. Ferrer, R. Deans, A. Moseley, R. Hoffman and S. M. Devine, Hum. Gene Ther., 2001, 12, 1527 CrossRef CAS.
- R. Kazlauskas, C. J. Howe and G. J. Trout, Clin. J. Sports Med., 2002, 12, 229 Search PubMed.
- G. Baumann, Endocr. Rev., 1991, 12, 424 Search PubMed.
- A. Breidbach and D. H. Catlin, Rapid Commun. Mass Spectrom., 2001, 15, 2379 CrossRef.
- T. M. S. Chang, Trends Biotechnol., 1999, 17, 61 CrossRef CAS.
- G. S. Hughes, Jr., E. P. Yancey, R. Albrecht, P. K. Locker, S. F. Francom, E. P. Orringer, E. J. Antal and E. E. Jacobs Jr., Clin. Pharm. Ther., 1995, 58, 434 CAS.
- H. F. Bunn, Transfus. Clin. Biol., 1995, 6, 443.
- C. W. Alma, G. J. Trout, N. Woodland and R. Kazlauskas, Recent Advances in Doping Analysis (10),W. Schänzer,H. Geyer, A. Gotzmann and U. Mareck-Engelke (eds.), Sport und Buch Strauss, Cologne, 2002, p. 169 Search PubMed.
- D. H. Catlin, B. D. Ahrens and Y. Kucherova, Rapid Commun. Mass Spectrom., 2002, 16(13), 1273 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2004 |