The photophysical properties of a pyrene–thiophene–terpyridine conjugate and of its zinc(II) and ruthenium(II) complexes
Andrew C.
Benniston
,
Anthony
Harriman
*,
Donald J.
Lawrie
and
Annabelle
Mayeux
Molecular Photonics Laboratory, School of Natural Sciences - Chemistry, Bedson Building, University of Newcastle, Newcastle upon Tyne, NE1 7RU, UK. E-mail: anthony.harriman@ncl.ac.uk; Fax: +44 (0)191 222 8664; Tel: +44 (0)191 222 8660
First published on 2nd December 2003
Abstract
A tripartite supermolecule, comprising a pyrene moiety tethered to a 2,2′:6′,2″-terpyridyl ligand via a 2,5-diethynylated thiophene linker, has been synthesized. This compound is highly fluorescent in solution due to the formation of an intramolecular charge-transfer (CT) state. From consideration of the electrochemical properties, it is concluded that the CT state arises because of charge transfer from pyrene to the central thiophene-based unit. Formation of the CT state involves an increase in dipole moment of ca. 18.5 D. Phosphorescence was not observed but the intermediate population of the triplet state was confirmed by laser flash photolysis. Addition of Zn2+ cations results in a drastic decrease in the fluorescence yield while the absorption spectrum exhibits a pronounced red shift. It appears that the dipole is extended upon cation binding, with the zinc terpyridine terminal acting as the electron acceptor. Again, no phosphorescence was apparent at 77 K. Coordination of a ruthenium(II) 2,2′;6′,2″-terpyridyl metallo-fragment to the vacant terpyridine terminal causes the appearance of weak phosphorescence in fluid solution at room temperature. The emitting species has a lifetime of 2.6 μs in deoxygenated acetonitrile at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C. Luminescence, which shows a complex temperature dependence, is attributed to either the lowest-energy metal-to-ligand, charge-transfer triplet localised on the ruthenium(II) complex or to the intraligand CT state. In the later case, spin–orbit coupling effects induced by the ruthenium atom are responsible for promoting emission.
°C. Luminescence, which shows a complex temperature dependence, is attributed to either the lowest-energy metal-to-ligand, charge-transfer triplet localised on the ruthenium(II) complex or to the intraligand CT state. In the later case, spin–orbit coupling effects induced by the ruthenium atom are responsible for promoting emission.
Introduction
In searching for new molecular systems able to function as fluorescence reporters for selected cations attention has focussed on highly conjugated supermolecules comprising an aryl hydrocarbon and a chelating ligand linked by an unsaturated connector.1 Prototypic among such sensors are pyrene–connector–poly(pyridine) units.2 Here, the poly(pyridine) terminal functions as a general purpose trap for coordinating adventitious cations and pyrene is intended as the fluorescent register. The connector must provide the necessary basis by which complexation of a cation modulates the fluorescence yield.3 Among the most effective fluorescence sensors are those where the free ligand forms an intramolecular charge-transfer (CT) state in polar solution.4 For such systems, coordination of a cation alters the internal electronic system in such a way that the CT state is rendered non-fluorescent. Several promising systems have now been developed along these lines.5A special case exists where the coordinated cation can form a luminescent complex. In particular, d6 cations such as ruthenium(II) or osmium(II) are known to form emissive complexes with poly(pyridine) ligands.6 Such systems often phosphoresce in fluid solution at ambient temperature. For these metal complexes,7 it appears that the CT triplet state associated with the ligand possesses a comparable energy to the metal-to-ligand, charge-transfer (MLCT) triplet state localised on the metal complex. Indeed, in at least one case these two excited states reside in thermal equilibrium at room temperature and give rise to dual emission.8
We recently described a general purpose chemical sensor built by connecting pyrene to 2,2′:6′,2″-terpyridine via a 2,5-diethynylene–thiophene unit.5b The free ligand was shown to be highly fluorescent in solution and to register complexation of many different cations, especially zinc(II) ions. This ligand forms a stable complex with ruthenium(II), after capping the metallo-fragment with a second 2,2′:6′,2″-terpyridine unit. The resultant metal complex emits weakly in fluid solution at room temperature, whereas the corresponding zinc(II) complex shows fluorescence but no phosphorescence. Although the triplet excited state of the parent complex, [Ru(terpy)2]2+, is too short-lived to phosphoresce under these conditions,9 it is known that attaching an alkynylene group at the 4′-position switches-on the radiative process.10 Consequently, the identity of the emitting state in the new supermolecule is far from obvious. Because of the intense interest in incorporating metallo-fragments in electro-optical devices11 we have examined the photophysical properties of this new system in detail.
Experimental
All chemicals were purchased from Aldrich Chemicals Co. and were used as received. Solvents were dried by standard literature methods12 before being distilled and stored under nitrogen over 4Å molecular sieves. 1H and 13C NMR spectra were recorded with a JEOL Lambda 500 spectrometer. Routine mass spectra and elemental analyses were obtained using in-house facilities. The free ligand L was prepared and purified as described earlier.5b The corresponding zinc(II) complex was prepared by adding a slight molar excess of zinc perchlorate to a solution of L in acetonitrile. Electrospray mass spectrometry was used to monitor the course of reaction and to conclude that the predominate species present under these conditions is the 1∶1 complex, ZnL.The corresponding ruthenium(II) complex, RuL, was prepared as follows (Scheme 1): to a refluxing solution of [Ru(terpy)(CH3CN)3)][PF6]2
(28 mg, 3.4×10−5 mol) in acetone (40 ml) and ethanol (20 ml) was added dropwise L (20 mg, 3.75×10−5 mol) in a solution of acetone (60 ml), ethanol (20 ml) and chloroform (10 ml). The resultant solution was refluxed at 95°C for six days before being cooled to room temperature. The solvent was evaporated to approximately 10 ml. A 10 ml aliquot of KPF6 solution (0.1 mol dm−3) was added to afford a red precipitate. The precipitate was filtered off and washed with water (50 ml), cold ethanol (25 ml) and diethyl ether (100 ml). The solid was purified by column chromatography (basic alumina, chloroform–acetonitrile (0 to 40%)) to yield 20 mg (49%) of the desired product as a red powder. This was further purified by vapour diffusion crystallisation (acetonitrile: diethyl ether) to give red crystals. 1H NMR (500 MHz, CDCl3): δ 1.33 (t, 3H, J=7.5 Hz, CH3), 2.92 (q, 2H, J=7.6 Hz, CH2), 6.21 (m, 4H, CHterpy), 7.53 (s, 1H, CHthio), 7.63 (dd, 2H, J=5.1 Hz, J′=0.6 Hz, CHterpy), 7.71 (dd, 2H, J=5.2 Hz, J′=0.6 Hz, CHterpy), 7.97 (qd, 4H, J=8.1 Hz, J′=1.2 Hz, CHterpy), 8.12 (m, 8H, CHpyrene), 8.49 (t, 1H, J=8.2 Hz, CHterpy), 8.53 (d, 1H, J=9.1 Hz, CHpyrene), 8.70 (d, 2H, J=8.3 Hz, CHterpy), 8.80 (d, 2H, J=7.9 Hz, CHterpy), 8.97 (d, 2H, J=8.3 Hz, CHterpy), 9.12 (s, 2H, CHterpy).13C NMR (125.65 MHz, CDCl3): δ 15.12, 23.80, 88.41, 89.82, 94.79, 95.13, 117.15, 118.32, 124.90 (2C), 125.16 (2C), 125.52 (2C), 125.72 (4C), 126.59, 127.08 (2C), 127.16, 127.70, 128.16, 128.65 (2C), 128.89 (2C), 129.77, 129.98, 130.50, 130.55, 131.93, 132.24, 132.67, 132.92, 134.50, 137.42, 139.20 (2C), 139.31 (2C), 153.36 (2C), 153.51 (2C), 153.60 (2C), 156.24 (2C), 156.59 (2C), 158.78 (2C), 159.11 (2C). ESI-MS values for C56H36N6SRuP2F12; m/z=1071.2 for M−[PF6], m/z=463.0 for M−2[PF6].
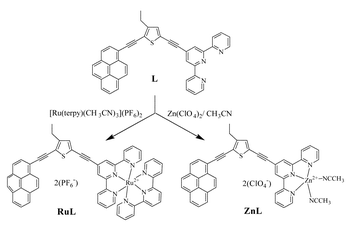 | ||
| Scheme 1 Structures of the compounds studied herein and their synthesis from the free ligand L. | ||
Cyclic voltammetry experiments were performed using a fully automated HCH Instruments Electrochemical Analyzer, and a three electrode set-up consisting of a glassy carbon working electrode, a platinum wire counter electrode and an Ag/AgCl reference electrode. All studies were performed in deoxygenated acetonitrile containing tetra-n-butylammonium tetrafluoroborate (0.2 mol dm−3) as background electrolyte. Reduction potentials were reproducible to within ±15 mV.
Absorption spectra were recorded with an Hitachi U3310 spectrophotometer while corrected emission spectra were recorded with an Hitachi F4500 spectrophotometer. All luminescence measurements were made using optically dilute solutions and were corrected for spectral imperfections of the instrument by reference to a standard lamp. Quantum yields were determined relative to either 9,10-diphenylanthracene in benzene13 or ruthenium(II) tris(2,2′-bipyridine) in acetonitrile.14 Time-resolved emission studies were made with a Spex Fluorolog tau-3 spectrophotometer while longer lifetimes were measured after excitation with a 4-ns laser pulse at 532 nm. Temperature dependence studies were made with an Oxford Instruments Optistat DN cryostat.
Transient absorption measurements were made with a Q-switched, frequency-doubled, Nd-YAG laser (FWHM=4 ns, λ=532 nm). For studies made with L or ZnL, output from the laser was frequency-tripled to provide excitation at 355 nm. Metal screen filters were used to attenuate the laser beam. The solution was deoxygenated by purging with argon and the monitoring beam was provided with a pulsed, high-intensity xenon arc lamp. The signal was directed to a high-radiance monochromator and then to a fast response transient digitiser. Transient differential absorption spectra were recorded point-by-point with five individual records being averaged at each wavelength. Kinetic measurements were made by averaging about 50 individual traces.
Spectral analysis were made using PeakFit to deconvolute the spectrum, displayed as intensity vs wavenumber, into the minimum number of Gaussian components. The latter parameter was decided on the basis of the residuals and R factor. The intensity was either the molar absorption coefficient or the reduced emission intensity (L(ν)=IF(ν)/ν3), where IF refers to the emission intensity at wavenumber ν.
Results and discussion
Cyclic voltammetry
Cyclic voltammograms recorded for L in deoxygenated acetonitrile containing tetra-n-butylammonium tetrafluoroborate (0.2 mol dm−3) as background electrolyte showed an irreversible peak at 1.31 V vs. Ag/AgCl. This process is most likely due to one-electron oxidation of the pyrene subunit.15 Oxidation of the thiophene unit in L can be observed at ca. 2.0 V vs. Ag/AgCl, although this is very close to the onset of solvent oxidation. On reductive scans, L exhibits a poorly reversible (ΔEp=140 mV) wave with E½=−1.61 V vs. Ag/AgCl, while at more cathodic potentials, a second reduction process occurs with E½=−1.90 V vs. Ag/AgCl. A tentative explanation for the reductive processes seen for L assigns the first step to the one-electron reduction of the central thiophene residue.16 The second reductive step corresponds to one-electron reduction of the terpyridyl terminal. Addition of a slight excess of Zn(ClO4)2 to a solution of L causes the appearance of a broad, quasi-reversible peak centred at −1.28 V vs. Ag/AgCl. This peak corresponds to two overlapping reduction steps, each of which is due to the transfer of one electron. The first reductive step can be attributed to reduction of the metal complex while the second step is due to reduction of the central thiophene unit. There is extensive literature evidence to indicate that coordination of a cation has a marked effect on the reduction potential of poly(pyridines).17,18 As such, we can surmise that complexation of Zn(ClO4)2 pushes the half-wave potential for reduction of the terpyridine unit to a much less negative value. The two overlapping peaks, therefore, can be assigned to one-electron reduction of the coordinated terpyridyl terminal and the central thiophene unit. The half-wave potential for this latter step is less negative by ca. 200 mV than that found in the absence of Zn2+ cations. This effect can be explained in terms of a cation-induced charge-shift reaction. Zinc(II) binding has a more pronounced effect on the half-wave potential for the terminal terpyridine unit but this is a well known effect. It should be noted that attachment of the cation has little effect on the oxidative processes.
Cyclic voltammograms recorded for RuL in deoxygenated acetonitrile solution indicate a two-electron oxidation process with the average peak position at 1.36 V vs. Ag/AgCl. This step corresponds to a combination of an irreversible oxidative process and a quasi-reversible electrode reaction. As such, we can assign this peak to the simultaneous oxidation of the pyrene and metal complex terminii. The central thiophene unit undergoes an irreversible oxidative process at 1.52 V vs. Ag/AgCl.
On reductive scans, quasi-reversible processes are observed with half-wave potentials of −1.07 and −1.35 V vs. Ag/AgCl. Both steps are assigned to the coordinated terpyridine ligands but the most easily reduced ligand is the one bearing the ethynylene substituent. In line with previous work, attachment of the ruthenium(II) centre renders the terpyridine ligand much easier to reduce.18,19 Even relative to the zinc(II) complex, the substituted ligand is easier to reduce by some 200 mV. There is now a substantial driving force (ΔG°≈−0.45 eV) for charge shift from the central thiophene unit to the terminal metal complex.
Photophysical properties of the free ligand
The absorption spectrum recorded for L in acetonitrile solution shows a series of transitions between 200 and 450 nm. The lowest-energy peak is observed at 426 nm, for which the molar absorption coefficient is 19550 mmol−1 cm2
(Fig. 1). However, there is an obvious shoulder on the low-energy side of this transition that disappears in non-polar solvent. This latter absorption band has the general appearance of a charge-transfer transition.20 According to our interpretation of the cyclic voltammograms, the electron donor is the pyrene terminal whilst the electron acceptor will be the central thiophene residue. Of course, there is extensive electron delocalisation over these subunits such that the exact nature of the donor and acceptor cannot be properly defined.
 | ||
| Fig. 1 Absorption spectra recorded for (a) L, (b) ZnL and (c) RuL in dilute acetonitrile solution. | ||
The absorption spectrum recorded in acetonitrile can be split into a series of Gaussian-shaped components. Under such conditions, the CT transition appears as a broad band (FWHM=2325 cm−1) centred at 22875 cm−1. The pyrene-like 1La transition21 appears as a series of three vibronic bands, with the first peak centred at 23443 cm−1. This latter band is much more intense than the CT band but there is considerable overlap between the first two absorption transitions. The spectrum shows the presence of other CT bands, centred at 26595 and 31345 cm−1 and an additional transition at 29500 cm−1. The oscillator strength (f) was calculated for the CT and 1La bands, using the measured molar absorption coefficient (ε) and allowing for the Gaussian analysis.22 The derived values are 0.016 and 0.17, respectively, for the CT and 1La bands. Because of the close spacing of these two absorption transitions, it is likely that they will interact, subject to symmetry rules. Such interaction might be expected to enhance radiative processes.23
In acetonitrile solution at ambient temperature, L displays intense fluorescence centred around 488 nm (Fig. 2). The fluorescence quantum yield is 0.32±0.03 while the fluorescence lifetime is 1.2±0.1 ns. The fluorescence intensity was unaffected by the presence of molecular oxygen, presumably because of the relatively short excited state lifetime. Comparison of absorption and emission spectral profiles indicates that there is a substantial Stokes' shift (i.e., ca. 62 nm) in acetonitrile. This finding is consistent with fluorescence arising from a charge-transfer state. The fluorescence excitation spectrum matches the absorption profile over the range 200 to 450 nm. The fluorescence spectrum is broad and fairly structureless. It does not resemble a mirror image of the lowest-energy absorption band. Taken together with the large Stokes's shift, this finding suggests to us that there is a significant geometry change after excitation.24
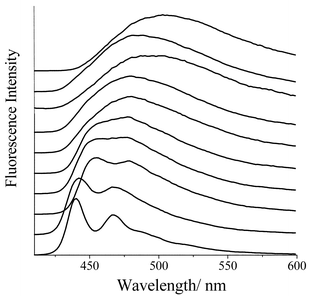 | ||
| Fig. 2 Fluorescence spectra recorded for L in solvents of differing polarity. From top to bottom, the solvents are dimethyl sulfoxide, acetonitrile, methanol, ethanol, butyronitrile, dichloromethane, tetrahydrofuran, chloroform, diethyl ether and cyclohexane. | ||
As shown in Fig. 2, the fluorescence profile depends on solvent polarity, although the lifetime remains around 1 ns. In non-polar solvents, the fluorescence spectrum is somewhat structured and shows only a modest Stokes shift. In cyclohexane, however, the fluorescence quantum yield is increased to 0.91 but the lifetime is only 0.96 ns. With increasing solvent polarity, the spectrum broadens and loses all semblance of fine structure. The Stokes shift increases progressively with solvent polarity and exhibits a linear dependence on the solvent polarity function, fS.25 According to the Lippert–Mataga expression,26 the solvent dependence indicates an increase in dipole moment of ca. 18.5±1.2 D for the excited state relative to the ground state. In calculating the dipole moment, the charge transfer distance is taken to be 7 Å, which corresponds to the distance between the centres of the pyrene and thiophene units.
In acetonitrile at 20°C, the radiative rate constant (kRAD)22 is 2.7×108 s−1 while the total reorganisation energy (λT) associated with relaxation of the Franck–Condon state to the emissive state is 1500 cm−1. The emission spectrum is well represented by three Gaussian peaks of common half-width (FWHM=2345 cm−1) The position of the highest-energy Gaussian (νF=21140 cm−1) corresponds to the 0,0 transition for the luminescence process. From the peak half-width, the total reorganisation accompanying charge-recombination fluorescence is calculated as 2400 cm−1. The average spacing between adjacent Gaussian peaks (hωM=1,850 cm−1) is assumed to represent a single medium-frequency vibrational mode coupled to deactivation of the excited state.
Using these parameters, the charge-recombination emission spectrum, presented in terms of intensity (L) vs. wavenumber (ν), could be reconstituted on the basis of eqn. (1).27
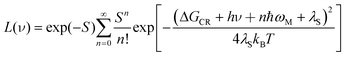 | (1) |
=23145 cm−1) or from the spectroscopic terms (ΔGCR=νF+λT=23540 cm−1). Both approaches give the same value, within experimental error. This analysis allows further calculation of S=0.48, λN=900 cm−1 and λS=1,500 cm−1. These values appear to be quite reasonable and in line with data collected for related CT states.28
The radiative rate constant can be expressed in terms of eqn. (2), which allows for coupling between the CT and 1La transitions.29
 | (2) |
20490 cm−1) refers to the peak maximum of the reduced emission spectrum and νA*
(=23445 cm−1) is the energy of the 0,0 band for the S0→1La transition. The term μ* represents the transition moment for the pyrene-like excited state and V* is the electronic coupling matrix element that describes mixing between the two excited states. The transition moment can be calculated from absorption spectral data:29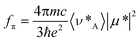 | (3) |
±0.2 D. The calculated matrix element (V*=4600±300 cm−1) indicates very strong coupling between the CT and 1La states. This state mixing is responsible for the high radiative rate constant found for L.
No phosphorescence could be detected for L in an ethanol glass at 77 K. However, laser flash photolysis studies clearly indicated formation of the triplet excited state in deoxygenated acetonitrile at room temperature. The transient differential absorption spectrum (Fig. 3) shows bleaching in the near UV region and broad absorption over the far red region. The signal decayed via first-order kinetics with an averaged lifetime of 35±5 μs in the absence of oxygen.
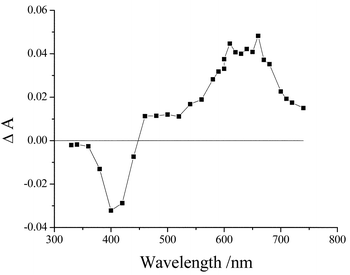 | ||
| Fig. 3 Triplet differential absorption spectrum recorded for L in deoxygenated acetonitrile solution following excitation with a 4-ns laser pulse at 355 nm. The spectrum was recorded 1 μs after the laser pulse. | ||
There are numerous examples of intramolecular CT states that undergo fast intersystem crossing to the triplet state in competition to non-radiative deactivation to the ground state.30 Such behaviour can be explained in terms of the energy-gap law31 since internal conversion demands the dissipation of a large amount of energy. In contrast, intersystem crossing requires the transfer of a much smaller amount of energy to the solvent bath and is therefore favoured.
Photophysical properties of ZnL
Addition of Zn(ClO4)2 to a solution of L affects the absorption spectrum in that the lowest-energy transitions are shifted towards longer wavelength and lose some of their fine structure (Fig. 1). This red shift is accompanied by a substantial increase in absorptivity over the 400–500 nm range. The lowest-energy absorption band, which has a molar absorption coefficient at 440 nm of 36035 mmol−1 cm2, can be approximated to a Gaussian-shaped profile with a maximum at 437 nm and a half-width of 2930 cm−1. As such, it seems reasonable to suppose that this absorption band corresponds to an intramolecular charge-transfer transition. The most reasonable interpretation of the experimental data has pyrene as the electron donor and the metal complex as the ultimate electron acceptor. Thus, the effect of cation coordination is to shift the charge from the central thiophene unit to the terminal metal complex. According to electrospray mass spectrometry, the dominate species under these conditions is ZnL.
In acetonitrile at room temperature, ZnL does not fluoresce within the spectral range 450 to 800 nm. However, the 1∶2 (metal∶ligand) complex, ZnL2, fluoresces very weakly with a maximum around 575 (Fig. 4). This latter emission band is independent of excitation wavelength. Furthermore, the corrected excitation spectrum agrees well with the absorption spectrum recorded over the 300–500 nm range. Under these conditions, the fluorescence quantum yield is ca. 0.006 but no low-temperature phosphorescence could be detected at λ<750 nm.
 | ||
| Fig. 4 Emission spectra recorded for ZnL2 (peak at 575 nm) and RuL (peak at 710 nm) in acetonitrile solution at room temperature. | ||
It seems likely that the excited singlet state of ZnL undergoes rapid non-radiative deactivation. Such processes might be promoted by the bound solvent molecules, as happens for many transition metal complexes.32 In particular, it should be stressed that neither ZnL nor ZnL2 phosphoresces at 77 K. Transient formation of the triplet state was confirmed by laser flash photolysis studies carried out with ZnL in deoxygenated acetonitrile at room temperature. The derived differential absorption spectrum (Fig. 5) differs sufficiently from that recorded for L for it to be assigned to the metal complex. The signal decays via first-order kinetics with an averaged lifetime of 43±7 μs in the absence of oxygen. It appears, however, that the triplet state is formed in quite low yield.
 | ||
| Fig. 5 Triplet differential absorption spectrum recorded for ZnL in deoxygenated acetonitrile solution following excitation with a 4-ns laser pulse at 355 nm. The spectrum was recorded 1 μs after the laser pulse. | ||
Photophysical properties of RuL
The absorption spectrum recorded for RuL shows the expected33 MLCT transition centred around 500 nm (Fig. 1). This absorption band is relatively intense, possessing a molar absorption coefficient of ca. 25000 mmol−1 cm2 at the peak. The presence of the parent terpyridine ligand is clearly evident from the absorption peak seen at 270 nm. Interestingly, the CT bands associated with the substituted ligand appear between 350 and 450 nm. These transitions closely resemble those found for L, rather than for ZnL. This suggests that the principal intraligand CT absorption transitions for RuL involve charge transfer from pyrene to the central thiophene unit. There are, however, new absorption bands in the near UV, with prominent transitions at 315 and 355 nm. These latter bands are characteristic of RuL and are intense. They cannot be assigned to the ethynylated terpyridine unit, since this group is also present in ZnL and ZnL2.
Linear extrapolation of the position of the intraligand CT absorption bands observed for L and ZnL, using the measured reduction potentials, suggests that the pyrene-to-terpyridine CT band for RuL should be centred around 455 nm. There is absorption in this region but it is masked by the more intense MLCT absorption transitions associated with the metal complex. We would expect to observe absorption bands at much higher energy that correspond to CT from thiophene to the coordinated terpyridine ligand and from the metal centre to the central thiophene unit.
A set of laser flash photolysis studies was made following excitation of RuL in deoxygenated acetonitrile with a 4-ns pulse at 532 nm. This wavelength corresponds to absorption solely by the metal complex. The resultant transient differential absorption spectrum (Fig. 6) shows strong bleaching of the MLCT transition centred at 500 nm and of the intraligand CT bands around 350–400 nm. There is also pronounced absorbance in the far-red region, with a peak around 830 nm. The spectrum is distinct from those recorded for both L and ZnL. Decay of the transient signal is independent of wavelength and involves first-order kinetics. The averaged lifetime is 2.6±0.2 μs. Molecular oxygen quenches the transient signal, suggesting that it arises from a triplet excited state. It is not possible, however, to identify the nature of the triplet state solely on the basis of the spectral profile.
 | ||
| Fig. 6 Triplet differential absorption spectrum recorded for RuL in deoxygenated acetonitrile solution following excitation with a 4-ns laser pulse at 532 nm. The spectrum was recorded 100 ns after the laser pulse. | ||
In deoxygenated butyronitrile at room temperature, RuL shows weak emission (Fig. 4). The luminescence peak is centred at 710 nm but whereas the lifetime (τL=2.5±0.2 μs) is quite long the quantum yield (ΦL=0.003±0.001) is relatively low. Good agreement exists between the corrected excitation spectrum and the absorption spectrum over the entire visible range. The long lifetime, together with the observation that the emission is extensively quenched by molecular oxygen, indicates that luminescence arises from a triplet excited state. Furthermore, the lifetime agrees well with that recorded by transient absorption spectroscopy. Thus, we conclude that the emitting species is the lowest-energy triplet state. The radiative rate constant (kRAD=ΦL/τL≈103 s−1) is significantly lower than expected for emission from an MLCT triplet state.34 Also, the peak maximum is red shifted with respect to related MLCT states where the luminophore is part of a conjugated supermolecule.16a,35 Such observations, while being far from definitive, are inconsistent with the observed emission arising from the MLCT triplet state localised on the metal complex.
The luminescence spectral profile and yield recorded for RuL in deoxygenated butyronitrile depend on temperature over the range 298 to 77 K. Thus, in fluid solution, the emission yield decreases progressively with increasing temperature between 170 and 298 K. An isosbestic point is preserved at 695 nm from 170 to 260 K but is lost at higher temperature. The emission maximum is located at 715 nm at the lower temperatures but shifts towards 710 nm as room temperature is approached. Over the entire temperature range, changes in the emission lifetime exactly parallel variations in the quantum yield.
Below the glass transition temperature, the emission maximum is blue-shifted to 702 nm. There is a 10-fold increase in emission yield and lifetime relative to room temperature but no real change in the spectral profile. In particular, the spectrum does not exhibit fine structure typical of emission from a π,π* triplet state.21
Spectral analysis36 of the emission profile recorded in a low-temperature glass indicates that the entire spectrum can be accounted for by two Gaussian bands. The highest energy band (FWHM=660 cm−1) is centred at 14720 cm−1 and is less intense than the lower energy band, which is centred at 14200 cm−1. The energy gap is only 520 cm−1. Assuming both bands arise from the same excited state, the energy gap indicates that a low frequency vibrational mode is coupled to the deactivation process. The total reorganisation energy associated with triplet decay at 113 K is then calculated to be 1000 cm−1.
An alternative explanation is that there are two emitting states in thermal equilibrium,37 even at low temperature. Analysing the emission spectra over a wide temperature range suggests that the latter situation is the more appropriate. Thus, the emission spectrum can be reasonably approximated to the sum of two Gaussian bands between 77 and 290 K. In frozen media the two bands are as described above but in fluid solution both bands move towards lower energy. Emission maxima are located at 14575 and 13970 cm−1, above the glass transition temperature. This corresponds to an approximate energy gap between the two states of 605 cm−1. As expected, the relative proportion of the higher energy band increases slightly with increasing temperature in both fluid and frozen solutions.
It is not possible to be definite about the identity of these two emitting excited states. However, the indications are that the lowest-energy triplet state corresponds to intraligand CT from pyrene to the coordinated terpyridine unit.3a,5b This ILCT state is considered to be responsible for the emission centred at 710–715 nm. The higher-energy triplet state is most likely the MLCT triplet localised on the metal complex. The energy gap (ΔE1=605 cm−1) is such that these two triplets will be in thermal equilibrium, with the relative population of the MLCT triplet (α) being set entirely by the temperature (T).37
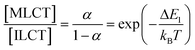 | (4) |
| k=αkMC+(1−α)kIC | (5) |
 | (6) |
 | ||
| Scheme 2 Proposed energy level diagram for the triplet state manifold of RuL. Both the MLCT and ILCT triplets emit. | ||
Analysis of the experimental data collected in fluid solution between 170 and 300 K provides reasonable parameters. Thus, the temperature-independent rate of decay of the ILCT triplet state (kIC) is derived to be (5.4±0.2)×104 s−1 while the temperature-independent rate of decay of the triplet MLCT state (k0) is found to be (2.4±0.5)×106 s−1
(Scheme 2). The energy gap between the MLCT triplet and the so-called dark state is calculated to be 2480±200 cm−1. Deactivation of the dark state, presumably directly to the ground state, occurs with a rate constant of (2.8±1.1)×1011 s−1. The identity of the dark state cannot be ascerted from the kinetic analysis but it is known that the pyrene-like triplet state has a relevant energy level.21 An alternative species is the metal-centred state that is believed to shorten the triplet lifetime of the parent ruthenium(II) bis(terpyridine) complex at ambient temperature.38
Effect of the central thiophene unit
Earlier work focussed on the photophysical properties of a pyrene-2,2′-bipyridine dyad (PB) and of a pyrene-ruthenium(II) bis(2,2′:6′,2″-terpyridine) (PTR) dyad, in both cases the terminals being connected via a single ethynylene group.3a The simple dyad PB behaves similarly to L and displays intense charge-transfer fluorescence in polar solvents. Relative to L, the emission maximum is blue-shifted by ca. 13 nm while the quantum yield and lifetime, respectively, are 0.025 and 3.0 ns. The change in dipole moment upon excitation is 24.4 D and it is clear that deactivation of the excited singlet state leads to population of the triplet state. This latter species has a lifetime of 130 μs in deoxygenated acetonitrile. Weak phosphorescence could be detected around 650 nm in a frozen glass at 77 K.Comparing PB with L indicates that the central thiophene unit promotes nonradiative deactivation of both excited singlet and triplet states. Very strong coupling between the CT and pyrene-like excited singlet state is observed for L. This state mixing must introduce considerable “singlet” character into the CT state. Such coupling provides for a very high radiative rate constant.29 Both L and PB undergo intersystem crossing to form the excited triplet state. Unfortunately, we were unable to detect low temperature phosphorescence from L but its triplet energy is unlikely to be higher than that found3a for PB (ET≈15380 cm−1). This latter value, which can be compared with the triplet energy of pyrene (ET≈16775 cm−1),21 sets the upper limit for the triplet energy of the free ligand L.
In acetonitrile solution, PTR shows a clear MLCT transition centred at 511 nm,16a which is red shifted with respect to RuL. The triplet excited state shows strong absorption around 800 nm, not unlike that found for RuL, but the emission peak is narrow and highly reminescent of that from a triplet MLCT state. This emission is centred around 700 nm, while the luminescence quantum yield and lifetime, respectively, are 0.005 and 580 ns. All the indications16a point to emission being from a triplet MLCT state for PTR, at least at room temperature.
The main difference between RuL and PTR, therefore, concerns the apparent inversion of the triplet levels. For PTR, the lowest-energy triplet is the MLCT state whereas an ILCT triplet falls slightly below the MLCT state for RuL. The energy of the triplet MLCT is about 15000 cm−1 for both PTR and RuL. It was proposed previously16a that the lowest-energy ligand-based triplet state for PTR is located around 15400 cm−1, while that for RuL is around 14000 cm−1. This is in line with the measured reduction potentials for the coordinated terpy ligand. Thus, reduction of RuL is easier by about 150 meV. This difference, which must be due to the presence of the thiophene unit, pushes the ILCT triplet towards lower energy and accounts for the transposition of the energy levels.
Conclusions
The central diethynylated thiophene residue in L is electron affinic and functions as an electron acceptor for an intraligand CT state. Pyrene acts as the electron donor. The intramolecular CT excited state is highly fluorescent in polar solvents and is significantly more polar than the ground state. It seems unlikely that the terminal terpyridine unit contributes towards the CT state since it is difficult to reduce under these conditions. However, the reduction potential of the terpyridine unit becomes less negative upon coordination of a metal cation. This is evidenced by a red-shifted absorption band, a marked decrease in fluorescence and a modification of the cyclic voltammograms. Such changes can be utilised to design chemical sensors that respond to the presence of adventitious cations in solution.5bMore importantly, capping the terminal terpyridine unit in L with a RuII–terpyridine metallo-fragment produces a “V-shaped” supermolecule that phosphoresces in deoxygenated solution at room temperature. Such emission is quite rare. In the present case, the emission lifetime is 2.5 μs in deoxygenated acetonitrile at 20°C but the quantum yield is quite low. The luminescence maximum is located around 710 nm in both butyronitrile and acetonitrile. This is a very useful spectral range for luminescent labels for biomaterials. The long emission lifetime allows time gating while the central thiophene unit is readily functionalised so as to form an appropriate anchor. Thus, RuL might be a valuable bio-label.
Acknowledgements
We thank the EPSRC and the University of Newcastle for financial support of this work.References
-
(a) A. S. Klymchenko and A. P. Demchenko, J. Am. Chem. Soc., 2002, 124, 12372 CrossRef CAS;
(b) G. Hennrich, H. Sonnenschein and U. Resch-Genger, J. Am. Chem. Soc., 1999, 121, 5073 CrossRef CAS.
- (a) A.-J. Tong, A. Yamauchi, T. Hayashita, Z.-Y. Zhang, B. D. Smith and N. Termae, Anal. Chem., 2001, 73, 1530 CrossRef CAS; (b) J. S. Kim, O. J. Shon, J. A. Rim, S. K. Kim and J. Yoon, J. Org. Chem., 2002, 67, 2348 CrossRef CAS; (c) T. Jin, Chem. Commun., 1999, 2491 RSC; (d) A. Corma, M. S. Galletero, H. García, E. Palomares and F. Rey, Chem. Commun., 2002, 1100 RSC; (e) Y. Fujiwara, I. Okura, T. Miyashita and Y. Amao, Anal. Chim. Acta., 2002, 471, 25 CrossRef CAS; (f) L. J. D'Souza and U. Maitra, J. Org. Chem., 1996, 61, 9494 CrossRef CAS; (g) D. Y. Sakaki and B. E. Padilla, Chem. Commun., 1998, 1581 RSC.
- (a) A. Harriman, M. Hissler and R. Ziessel, Phys. Chem. Chem. Phys., 1999, 1, 4203 RSC; (b) L. Fabbrizzi, M. Licchelli, G. Rabaioli and A. Taglietti, Coord. Chem. Rev., 2000, 205, 85 CrossRef CAS; (c) J. Kawakami, Y. Komai, T. Sumori, A. Fukushi, K. Shimozaki and S. Ito, J. Photochem. Photobiol. A, Chem., 2001, 139, 71 CrossRef CAS.
-
(a) L. Fabbrizzi, M. Licchelli, P. Pallavicini and D. Sacchi, Supramol. Chem., 2001, 13, 569 CrossRef CAS;
(b) T. Gunnlaughsson, A. P. Davis and M. Glynn, Chem. Commun., 2001, 2556 RSC;
(c) K. Rurack and U. Resch-Genger, Chem. Soc. Rev., 2002, 31, 116 RSC;
(d) X. Zhang, C. J. Wang, L. H. Liu and Y. B. Jiang, J. Phys. Chem. B, 2002, 106, 12432 CAS;
(e) P. G. Cozzi, L. S. Dolci, A. Garelli, M. Montalti, L. Prodi and N. Zaccheroni, New J. Chem., 2003, 27, 692 RSC;
(f) P. Du, C. Li, S. F. Li, W. H. Zhu and H. Tian, Synth. Metals, 2003, 137, 1131 CrossRef CAS;
(g) M. Sauer, Angew. Chem., Int. Ed., 2003, 42, 1790 CrossRef CAS;
(h) A. Chatterjee, T. M. Suzuki, Y. Takahashi and D. A. P. Tanaka, Chem.–Eur. J., 2003, 9, 3920 CrossRef CAS;
(i) A. S. Klymchenko, A. S. Pivovarenko, T. Ozturk and A. P. Demchenko, New J. Chem., 2003, 27, 1336 RSC.
- (a) S. Leroy, T. Soujanya and F. Fages, Tetrahedron Lett., 2001, 42, 1665 CrossRef CAS; (b) A. C. Benniston, A. Harriman, D. J. Lawrie, A. Mayeux, K. Rafferty and O. D. Russell, Dalton Trans., 2003, 4762 RSC; (c) S. Aoki, S. Kaido, H. Fujioka and E. Kimura, Inorg. Chem., 2003, 42, 1023 CrossRef CAS.
- (a) J. V. Caspar, T. D. Westermoreland, G. H. Allen, P. G. Bradley, T. J. Meyer and W. H. Woodruff, J. Am. Chem. Soc., 1984, 106, 3492 CrossRef CAS; (b) L. Mishra, A. K. Yadaw and G. Govil, Indian J. Chem., Sect. A, 2003, 42, 1797.
- A. D. Guerzo, S. Leroy, F. Fages and R. H. Schmehl, Inorg. Chem., 2002, 41, 359 CrossRef CAS.
- For a detailed discussion of the literature see A. Harriman, A. Khatyr and R. Ziessel, J. Chem. Soc., Dalton Trans., 2003, 2061 Search PubMed.
- A. El-ghayoury, A. Harriman, A. Khatyr and R. Ziessel, Angew. Chem., Int. Ed., 2000, 39, 185 CrossRef CAS.
- A. Harriman and R. Ziessel, Coord. Chem. Rev., 1998, 171, 331 CrossRef.
- (a) T. Tsuboi and M. Tanigawa, Thin Solid Films, 2003, 438, 301 CrossRef; (b) E. Stathatos, P. Lianos and E. Evgeniou, Synth. Met., 2003, 139, 433 CrossRef CAS; (c) S. H. Lim, X. Gong and J. Ostrowski, Chem. Phys. Lett., 2003, 376, 55 CrossRef CAS; (d) T. S. Kang, B. S. Harrison and T. J. Foley, Adv. Mater., 2003, 15, 1093 CrossRef CAS; (e) S. W. Magennis, A. J. Ferguson and T. Bryden, Synth. Met., 2003, 138, 463 CrossRef CAS; (f) S. Welter, K. Brunner, J. W. Hofstraat and L. De Cola, Nature, 2003, 421, 54 CrossRef CAS.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Permagon Press Ltd., Oxford, 3rd edn., 1988 Search PubMed.
- T. Suzuki, M. Nagano, S. Watanabe and T. Ichimura, J. Photochem. Photobiol. A, Chem., 2000, 136, 7 CrossRef CAS.
- J. V. Caspar and T. J. Meyer, J. Am. Chem. Soc., 1983, 105, 5583 CrossRef CAS.
- J. L. Sessler, Y. Kubo and A. Harriman, J. Phys. Org. Chem., 1992, 5, 644 CAS.
- (a) M. Hissler, A. Harriman, A. Khatyr and R. Ziessel, Chem.–Eur. J., 1999, 5, 3366 CrossRef CAS; (b) L. M. Voger, S. W. Jones, G. E. Jensen, G. Brewer and K. J. Brewer, Inorg. Chim. Acta., 1996, 250, 155 CrossRef.
- (a) G. Albano, V. Balzani, E. C. Constable, M. Maestri and D. R. Smith, Inorg. Chim. Acta, 1998, 227, 227; (b) J. E. S. Sohna, P. Jaumier and F. Fages, J. Chem. Res. (S), 1999, 134 RSC; (c) K. R. J. Thomas, J. T. Lin, C. P. Chang, C. H. Chuen and C. C. Cheng, J. Chin. Chem. Soc., 2002, 49, 833 CAS.
-
(a) A. C. Benniston, V. Grosshenny, A. Harriman and R. Ziessel, Angew. Chem., Int. Ed., 1994, 33, 1884 CrossRef;
(b) V. Grosshenny, A. Harriman, J. P. Gisselbrecht and R. Ziessel, J. Am. Chem. Soc., 1996, 118, 10315 CrossRef CAS.
- M. Hissler, A. El-ghayoury, A. Harriman and R. Ziessel, Angew. Chem., Int. Ed., 1998, 37, 1711 CrossRef CAS.
- (a) N. Kitaguchi, Bull. Chem. Soc. Jpn., 1989, 62, 800 CAS; (b) K. Yamamura, K. Nakasuji, I. Murata and S. Inagaki, J. Chem. Soc., Chem. Commun., 1982, 396 RSC; (c) I. Murata, Pure Appl. Chem., 1983, 55, 323 CAS.
- J. B. Birks, Photophysics of Aromatic Molecules, Wiley-Interscience, New York, 1970 Search PubMed.
- S. J. Strickler and R. A. Berg, J. Chem. Phys., 1962, 37, 814 CrossRef CAS.
- I. R. Gould, R. H. Young, L. J. Meuller, A. C. Albrecht and S. Farid, J. Am. Chem. Soc., 1994, 116, 8188 CrossRef CAS.
- L. R. Khundkar, A. E. Stiegman and J. W. Perry, J. Phys. Chem., 1990, 94, 1224 CrossRef CAS.
- There is some discussion as to the most appropriate solvent function to use in the Lippert–Mataga expression. We used: fs=((ε−1)/(2ε+1))((n2−1)/(2n2+1)) where ε is the solvent dielectric constant and n is the refractive index.
- (a) E. Lippert, Z. Naturforsch., 1955, 10, 541 Search PubMed; (b) N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc, Jpn., 1955, 28, 690 CAS.
- M. Bixon, J. Jortner, J. Cortes, H. Heitele and M. E. Michel-Beyerle, J. Phys. Chem., 1994, 98, 7289 CrossRef CAS.
- J. Cortes, H. Heitele and J. Jortner, J. Phys. Chem., 1994, 98, 2527 CrossRef CAS.
- M. Bixon, J. Jortner and J. Verhoeven, J. Am. Chem. Soc., 1994, 116, 7349 CrossRef CAS.
- (a) M. R. Wasielewski, D. W. Minsek, M. P. Niemczyk, W. A. Svec and N. C. Yang, J. Am. Chem. Soc., 1990, 112, 2823 CrossRef CAS; (b) A. M. Brun, A. Harriman, Y. Tsuboi, T. Okada and N. Mataga, J. Chem. Soc., Faraday Trans., 1995, 91, 4047 RSC; (c) T. Asahi and N. Mataga, J. Phys. Chem., 1983, 93, 6575.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145 CAS.
- (a) A. E. Friedman, J.-C. Chambron, J.-P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1990, 112, 4960 CrossRef CAS; (b) P. Federlin, J.-M. Kern, A. Rastegar, C. Dietrich-Buchecker, P. A. Marnot and J.-P. Sauvage, New J. Chem., 1990, 14, 9 Search PubMed.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS.
- J.-P. Sauvage, J.-P. Collin, J.-C. Chambron, S. Guillerez, C. Coudret, V. Balzani, F. Barigelletti, L. De Cola and L. Flamigni, Chem. Rev., 1994, 94, 993 CrossRef CAS.
- (a) M. Hissler, A. El-ghayoury, A. Harriman and R. Ziessel, Coord. Chem. Rev., 1998, 178–80, 1251; (b) A. Harriman, M. Hissler, A. Khatyr and R. Ziessel, Eur. J. Inorg. Chem., 2003, 955 CrossRef CAS.
- Z. Murtaza, D. K. Graff, A. P. Zipp, L. A. Worl, W. E. Jones Jr., W. E. Bates and T. J. Meyer, J. Phys. Chem., 1994, 98, 10504 CAS.
- W. E. Ford and M. A. J. Rodgers, J. Phys. Chem., 1992, 96, 2917 CrossRef CAS.
- C. R. Hecker, A. K. I. Gushurst and D. R. McMillan, Inorg. Chem., 1991, 30, 538 CrossRef CAS.
| This journal is © the Owner Societies 2004 |